




General Information About Neuroblastoma
Dramatic improvements in survival have been achieved for children and adolescents with cancer.[1,2] Between 1975 and 2020, childhood cancer mortality decreased by more than 50%.[1,2,3,4,5] Between 1975 and 2017, the 5-year survival rate for patients with neuroblastoma increased, from 86% to 91% for children younger than 1 year and from 34% to 83% for children aged 1 to 14 years.[2,3]
Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. For specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
Incidence and Epidemiology
Neuroblastoma is the most common extracranial solid tumor in childhood. More than 650 cases are diagnosed each year in the United States.[2,6,7,8] The prevalence is about 1 case per 7,000 live births. The incidence is 8.3 cases per 1 million per year in children younger than 15 years. The overall incidence of neuroblastoma cases in the United States has remained stable.[9] About 37% of patients are diagnosed as infants, and 90% are younger than 5 years at diagnosis, with a median age at diagnosis of 17 months.[8,10] The data on age at diagnosis show that this is a disease of infancy, with the highest rate of diagnosis in the first month of life.[6,10,11]
Population-based studies of screening for infants with neuroblastoma have demonstrated that spontaneous regression of neuroblastoma without clinical detection in the first year of life is at least as prevalent as clinically detected neuroblastoma.[12,13,14]
The United States Cancer Statistics database and the National Program of Cancer Registries survival database was used to describe epidemiological trends in incidence and outcomes in patients with neuroblastoma between 2003 and 2019. Non-Hispanic White patients have a higher risk of developing neuroblastoma than all other race and ethnicity groups. Compared with non-Hispanic White patients, the relative risks were 0.54 for Hispanic patients, 0.64 for non-Hispanic Asian or Pacific Islander patients, 0.69 for non-Hispanic American Indian and Alaska Native patients, and 0.73 for non-Hispanic Black patients.[9] The 5-year relative survival rate was higher for non-Hispanic White patients (80.7%) or Hispanic patients (80.8%), compared with non-Hispanic Black patients (72.6%).[9]
Epidemiological studies have shown that environmental or other exposures have not been unequivocally associated with increased or decreased incidences of neuroblastoma.[15]
Anatomy
Neuroblastoma originates in the adrenal medulla and paraspinal or periaortic regions where sympathetic nervous system tissue is present (see Figure 1).
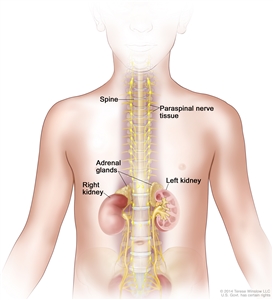
Figure 1. Neuroblastoma may be found in the adrenal glands and paraspinal nerve tissue from the neck to the pelvis.
Neuroblastoma Screening
Familial neuroblastoma and genetic predisposition
Studies analyzing constitutional DNA in rare cohorts of patients with familial neuroblastoma have provided insight into the complex genetic basis for tumor initiation. About 1% to 2% of patients with neuroblastoma have a family history of the disease. These children are, on average, younger (9 months at diagnosis) than patients without a family history, and about 20% of these patients have multifocal primary neuroblastoma.
Germline variants. Several germline variants have been associated with a genetic predisposition to neuroblastoma, including the following:
- ALK gene variant. The primary cause of familial neuroblastoma (about 75% of familial cases) is aberrant activation of the germline ALK signaling pathway, which results from single nucleotide variants in the tyrosine kinase domain of the ALK gene.[16] Somatic activating single nucleotide variants in ALK are also seen in about 9% of sporadic neuroblastoma cases. In addition, in a small proportion of neuroblastoma cases with MYCN amplification, ALK is co-amplified (ALK is near MYCN on chromosome 2), which may also result in ALK activation. ALK is a tyrosine kinase receptor. For more information about ALK variants, see the Genomic and Biological Features of Neuroblastoma section.
- PHOX2B gene variant. Rarely, familial neuroblastoma may be associated with congenital central hypoventilation syndrome (Ondine curse), which is caused by a germline variant of the PHOX2B gene.[17] Most PHOX2B variants causing Ondine curse or Hirschsprung disease are polyalanine repeats and are not associated with familial neuroblastoma. However, germline loss-of-function PHOX2B variants have been identified in rare patients with sporadic neuroblastoma and Ondine curse and/or Hirschsprung disease.[18] This aberration has not been seen in patients with sporadic neuroblastoma without associated Ondine curse or Hirschsprung disease. Additionally, somatic PHOX2B variants occur in about 2% of sporadic cases of neuroblastoma.[19,20]
- Deletion at the 1p36 or 11q14-23 locus. In case studies, germline deletion at the 1p36 or 11q14-23 locus has been associated with familial neuroblastoma. The same deletions are found somatically in some sporadic neuroblastoma cases.[21,22]
Other cancer predisposition syndromes. Children with gene aberrations associated with other cancer predisposition syndromes may be at increased risk of developing neuroblastoma and other malignancies. The following syndromes primarily involve genes in the canonical RAS pathway:
In addition, neuroblastoma has been described in patients with the following syndromes:
- Li-Fraumeni syndrome.
- Hereditary pheochromocytoma/paraganglioma syndromes.[26]
- ROHHAD syndrome (rapid-onset obesity, hypothalamic dysfunction, hypoventilation, and autonomic dysfunction).[27]
- Beckwith-Wiedemann syndrome.[28]
With increased availability of sequencing techniques, the spectrum of germline alterations seen in patients with neuroblastoma is expanding. For example, one study identified a series of 11 patients with germline pathogenic variants in SMARCA4.[29] In another study of 786 patients with neuroblastoma, 13.9% had pathogenic or likely pathogenic germline variants in cancer predisposition genes. BARD1, ERCC2, CHEK2, and MSH3 were the genes in which germline pathogenic variants were most commonly observed. Germline pathogenic variants in BARD1, EZH2, ALK, PTCH1, and MSH3 were specifically enriched in patients with neuroblastoma, compared with controls. Patients with these alterations had inferior survival, compared with patients without these alterations.[30] For more information about SMARCA4, visit Rhabdoid Tumor Predisposition Syndrome Type 2.
Sporadic neuroblastoma may also have an increased incidence resulting from less potent germline predispositions. Genome-wide association studies have identified several common genomic variables (single nucleotide polymorphisms) with modest effect size that are associated with increased risk of developing neuroblastoma. Most of these genomic risk variables are significantly associated with distinct neuroblastoma phenotypes (i.e., high-risk vs. low-risk disease).[31]
Neuroblastoma predisposition and surveillance
Screening recommendations from the American Association for Cancer Research (AACR) emerged from the 2016 Childhood Cancer Predisposition Workshop. The AACR recommends that the following individuals undergo biochemical and radiographic surveillance for early detection of tumors in the first 10 years of life:[26]
- Individuals with highly penetrant, heritable ALK or PHOX2B pathogenic variants (45%–50% risk of developing one or more tumors).
- Individuals with Li-Fraumeni syndrome and germline TP53 p.R337H pathogenic variants.
- Individuals with Beckwith-Wiedemann syndrome and germline CDKN1C pathogenic variants.
- Individuals with Costello syndrome and HRAS pathogenic variants.
- Individuals with neuroblastoma and a strong family history of neuroblastoma or clearly bilateral/multifocal neuroblastoma.
Surveillance consists of the following:[26]
- Abdominal ultrasonography.
- Quantitative, normalized assessment of urinary catecholamines,[32] such as urine vanillylmandelic acid (VMA) and homovanillic acid (HVA), by gas chromatography and mass spectroscopy (can be a random urine collection normalized for urine creatinine, because this approach appears to have similar sensitivity to a 24-hour collection).
- Chest x-ray.
Surveillance begins at birth or at diagnosis of neuroblastoma predisposition and continues every 3 months until age 6 years, then every 6 months until age 10 years. Patients with Costello syndrome may have elevated urinary catecholamines in the absence of a catecholamine-secreting tumor, so only high or significantly rising levels should prompt investigation beyond ultrasonography and chest x-ray.[33] Patients with Li-Fraumeni syndrome should not undergo chest x-rays.[26]
About 5% of children with Beckwith-Wiedemann syndrome have variants that cause decreased activity of CDKN1C. A review of all large studies of genetically subtyped Beckwith-Wiedemann syndrome found 70 children with the CDKN1C variant, 4.6% of whom developed neuroblastoma. There were no cases of Wilms tumor or hepatoblastoma. There is little experience with screening these children for neuroblastoma, so there are no generally accepted guidelines. However, the authors of the study suggest screening with urinary VMA/HVA every 4 to 6 months. Patients with other genetic subtypes of Beckwith-Wiedemann syndrome have a prevalence of neuroblastoma of less than 1%. No neuroblastic tumors were found among 123 children with the genotype gain of methylation at imprinting control region 1.[34]
General population
Current data do not support neuroblastoma screening in the general public. Screening at the ages of 3 weeks, 6 months, or 1 year did not lead to a reduced incidence of advanced-stage neuroblastoma with unfavorable biological characteristics in older children, nor did it reduce overall mortality from neuroblastoma.[13,14] No public health benefits have been shown from screening infants for neuroblastoma at these ages. For more information, see Neuroblastoma Screening.
Evidence (against neuroblastoma screening):
- In a large population-based North American study, most infants in Quebec, Canada, were screened at the ages of 3 weeks and 6 months.[12,13]
- Screening detected many neuroblastomas with favorable characteristics that would never have been detected clinically because of spontaneous regression of the tumors.
- Another study of infants screened at the age of 1 year showed similar results.[14]
Clinical Presentation
The most frequent signs and symptoms of neuroblastoma in children are caused by tumor mass and metastases and include the following:
- Abdominal mass: The most common presentation of neuroblastoma.
- Proptosis and periorbital ecchymosis: Common in high-risk patients; arise from retrobulbar metastasis.
- Abdominal distention: May occur with respiratory compromise in infants because of massive liver metastases.
- Bone pain: Occurs in association with metastatic disease.
- Pancytopenia: May result from extensive bone marrow metastasis.
- Fever, hypertension, and anemia: Occasionally found in patients without metastasis.
- Paralysis: Neuroblastoma originating in paraspinal ganglia may invade through neural foramina and compress the spinal cord extradurally. Immediate treatment is given for symptomatic spinal cord compression. For more information, see the Treatment of Spinal Cord Compression section.
- Watery diarrhea: On rare occasions, children may have severe, watery diarrhea caused by the secretion of vasoactive intestinal peptide by the tumor, or they may have protein-losing enteropathy with intestinal lymphangiectasia.[35] Vasoactive intestinal peptide secretion may occur at presentation (with diarrhea being the first symptom of neuroblastoma), may appear with the initiation of chemotherapy, or occasionally may become evident later in the course of treatment. Tumor resection reduces vasoactive intestinal peptide secretion.[36]
- Presence of Horner syndrome: Characterized by miosis, ptosis, and anhidrosis. It may be caused by neuroblastoma in the stellate ganglion. Children with Horner syndrome without other apparent causes are also examined for neuroblastoma and other tumors.[37]
- Subcutaneous skin nodules: Subcutaneous metastases of neuroblastoma often have bluish discoloration of the overlying skin; usually seen only in infants.
The clinical presentation of neuroblastoma in adolescents is similar to that in children. The only exception is that bone marrow involvement occurs less frequently in adolescents, and there is a greater frequency of metastases in unusual sites such as lung or brain.[38]
Opsoclonus/myoclonus syndrome
Paraneoplastic neurological findings, including cerebellar ataxia or opsoclonus/myoclonus, occur rarely in children with neuroblastoma.[39] Of young children presenting with opsoclonus/myoclonus syndrome, about one-half are found to have neuroblastoma.[40,41] The incidence in the United Kingdom is estimated at 0.18 cases per 1 million children per year. The average age at diagnosis is 1.5 to 2 years.[42]
The usual presentation is the onset of progressive neurological dysfunction over a few days before a neuroblastoma is discovered. However, on occasion, neurological symptoms arise long after removal of the primary tumor.[40,43,44] Patients with neuroblastoma who present with opsoclonus/myoclonus syndrome often have neuroblastoma with favorable biological features and have excellent survival rates, although tumor-related deaths have been reported.[40]
The opsoclonus/myoclonus syndrome appears to be caused by an immunologic mechanism that is not yet fully characterized.[40] The primary tumor is typically diffusely infiltrated with lymphocytes.[45] Cerebrospinal fluid shows an increased number of B cells, and oligoclonal immunoglobulin bands are often seen. Steroid-responsive elevations of B-cell–related cytokines are also often seen.[46]
Genomic copy number profiles were analyzed in 44 cases of neuroblastoma associated with opsoclonus/myoclonus syndrome. Because there were no tumor relapses or disease-related deaths, the overall genomic profile was not prognostically significant.[47]
Some patients may rapidly respond neurologically to immune interventions or simply to removal of the neuroblastoma, but in many cases, improvement may be slow and partial. The improvement in acutely presenting motor deficits and ataxia seen with immunological therapy is not clearly associated with improvement in long-term neuropsychological disability, which primarily consists of cognitive and behavioral deficits. The long-term benefits of rapid improvement resulting from treatment, whether of symptoms or of the underlying neuroblastoma, are unclear, but rapid improvement appears to be worthwhile.[44,48]
Treatment with adrenocorticotropic hormones or corticosteroids can be effective for acute symptoms, but some patients do not respond to corticosteroids.[43,49] Other therapy with various immunomodulatory drugs, plasmapheresis, intravenous gamma globulin, and rituximab have been reported to be effective in select cases.[43,50,51,52,53] Combination immunosuppressive therapy has been explored, with improved short-term results.[54] The short-term neurological outcomes may be superior in patients treated with chemotherapy, possibly because of its immunosuppressive effects.[39]
The Children's Oncology Group (COG) has completed the first randomized, open-label, phase III study of patients with opsoclonus/myoclonus ataxia syndrome.[55] Patients with newly diagnosed neuroblastoma and opsoclonus/myoclonus ataxia syndrome who were younger than 8 years were randomly assigned to receive either intravenous immunoglobulin (IVIG) or no IVIG in addition to prednisone and risk-adapted treatment of the tumor.[55]
- Of the 53 patients who participated, 21 of 26 patients (81%) in the IVIG group had an opsoclonus/myoclonus ataxia syndrome response over a period of weeks to months, compared with 11 of 27 patients (41%) in the non-IVIG group (odds ratio [OR], 6.1; P = .0029).
- This study demonstrated that short-term neurological response is improved in patients treated with chemotherapy, corticosteroids, and immunoglobulin, compared with patients treated with chemotherapy and corticosteroids without immunoglobulin.
- Patients on the trial were monitored to track adaptive (n = 25) and cognitive functioning (n = 15) over time. Both adaptive and cognitive functioning remained grossly stable during the first 2 years after diagnosis. Assessments beyond 2 years were limited by small sample sizes.[56] Additional data are needed to assess long-term neurodevelopmental and learning problems in this population.
Diagnosis
Diagnostic evaluation of neuroblastoma includes the following:
- Tumor imaging: Imaging of the primary tumor mass is generally accomplished by computed tomography or magnetic resonance imaging (MRI) with contrast. Paraspinal tumors that might threaten spinal cord compression are imaged using MRI.
Metaiodobenzylguanidine (MIBG) scanning is a critical part of the standard diagnostic evaluation of neuroblastoma, for both the primary tumor and sites of metastases.[57,58] MIBG scanning is also critical to assess response to therapy.[58] About 90% of neuroblastoma cases are MIBG avid. Fluorine F 18-fludeoxyglucose positron emission tomography (PET) scans are used to evaluate extent of disease in patients with tumors that are not MIBG avid.[59] For more information about imaging of neuroblastoma, see the Stage Information for Neuroblastoma section.
- Urine catecholamine metabolites: Urinary excretion of the catecholamine metabolites VMA and HVA per milligram of excreted creatinine is measured before therapy. Collection of urine for 24 hours is not needed. If they remain elevated, these markers can be used to suggest the persistence of disease.
In contrast to urine, serum catecholamines are not routinely used in the diagnosis of neuroblastoma except in unusual circumstances.
- Biopsy: Tumor tissue is often needed to obtain all the biological data required for risk-group assignment and subsequent treatment stratification in current COG clinical trials. There is an absolute requirement for tissue biopsy to determine the International Neuroblastoma Pathology Classification (INPC). Additionally, a significant number of tumor cells are needed to determine MYCN copy number, DNA index, and the presence of segmental chromosomal aberrations. Tissue from several core biopsies, or approximately 1 cm3 of tissue from an open biopsy, is needed for adequate biological staging. A systematic review of eight retrospective studies showed that both surgical biopsy and core-needle biopsy produced similar rates of obtaining adequate tissue for histopathological diagnosis and molecular characterization. Core-needle biopsy was associated with lower complication rates and reduced transfusion requirements.[60] Core-needle biopsy also appears to yield sufficient material for assessment of ALK status. In one single-center report of patients with neuroblastoma who were newly diagnosed using core-needle biopsy, ALK status was determined in 88% of cases.[61]
For patients older than 18 months with stage 4 disease, bone marrow with extensive tumor involvement combined with elevated catecholamine metabolites may be adequate for diagnosis and assigning risk/treatment group. However, INPC cannot be determined from tumor metastatic to bone marrow. Testing for MYCN amplification may be successfully performed on involved bone marrow if there is at least 30% tumor involvement. However, every attempt should be made to obtain an adequate biopsy from the primary tumor.
For information about the use of biopsy in patients younger than 1 year, see the Observation and Spontaneous Regression of Fetal/Neonatal Neuroblastoma section.
The diagnosis of neuroblastoma requires the involvement of pathologists who are familiar with childhood tumors. Some neuroblastomas cannot be differentiated morphologically, via conventional light microscopy with hematoxylin and eosin staining alone, from other small round blue cell tumors of childhood, such as lymphomas, Ewing sarcoma, and rhabdomyosarcomas. In such cases, immunohistochemical and cytogenetic analysis may be needed to diagnose a specific small round blue cell tumor.
The minimum criterion for a diagnosis of neuroblastoma, as established by international agreement, is that diagnosis must be based on one of the following:
- An unequivocal pathological diagnosis made from tumor tissue by light microscopy (with or without immunohistology or electron microscopy).[62]
- The combination of bone marrow aspirate or trephine biopsy containing unequivocal tumor cells (e.g., syncytia or immunocytologically positive clumps of cells) and increased levels of urinary catecholamine metabolites.[62]
Observation and Spontaneous Regression of Fetal/Neonatal Neuroblastoma
The phenomenon of spontaneous regression has been well described in infants with neuroblastoma, especially in infants with the INSS 4S/INRG MS pattern of metastatic spread.[63] In rare cases, neuroblastoma may be discovered prenatally by fetal ultrasonography.[64] Management recommendations are evolving regarding the need for immediate diagnostic biopsy in infants aged 6 months and younger with suspected neuroblastoma tumors that are likely to spontaneous regress. For more information about INSS 4S/INRG MS disease, see the Stage Information for Neuroblastoma section.
Spontaneous regression generally occurs in tumors with the following features:[65,66,67]
- Near triploid number of chromosomes.
- No MYCN amplification.
- No loss of chromosome 1p.
Additional features associated with spontaneous regression include the lack of telomerase expression,[65,68] the expression of the H-Ras protein,[69] and the expression of the neurotrophin receptor TrkA, a nerve growth factor receptor.[70]
Studies have suggested that selected infants who appear to have asymptomatic, small, low-stage adrenal neuroblastoma detected by screening or during prenatal or incidental ultrasonography often have tumors that spontaneously regress. These patients may be observed safely without surgical intervention or tissue diagnosis.[71,72,73]
Evidence (observation [spontaneous regression]):
- In a COG study, 83 highly selected infants younger than 6 months with stage 1 small adrenal masses (3.1 cm or less), as defined by imaging studies, were observed without biopsy. Surgical intervention was reserved for those with growth or progression of the mass or increasing concentrations of urinary catecholamine metabolites.[74]
- Eighty-one percent of patients did not undergo surgery, and all patients were alive after 2 years of follow-up. For more information, see the Surgery section.
- Therefore, prenatally identified adrenal masses approximately 3.1 cm or less can be safely observed if no metastatic disease is identified and there is no involvement of large vessels or organs.
- A German clinical trial reported on 340 infants with localized neuroblastoma without MYCN amplification. Of these patients, 190 underwent resection, 57 were treated with chemotherapy, and 93 were observed with gross residual tumor.[75]
- Spontaneous regression and/or lack of progression occurred in 44 of 93 asymptomatic infants aged 12 months or younger with stage 1, 2, or 3 tumors without MYCN amplification.
- All patients were observed after biopsy and partial or no resection.
- In some cases, regression did not occur until more than 1 year after diagnosis.
- In neuroblastoma screening trials in Quebec, Canada, and Germany, the incidence of neuroblastoma was twice that reported without screening, suggesting that many neuroblastomas are never diagnosed clinically and spontaneously regress.[12,13,14]
Prognostic Factors
The prognosis for patients with neuroblastoma is related to the following:
- Treatment era.
- Age at diagnosis.
- Tumor histology.
- Biological features.
- Site of primary tumor.
- Stage of disease.
- Response to treatment.
- Levels of lactate dehydrogenase (LDH) and ferritin.
Some of these prognostic factors have been combined to create risk groups to help define treatment. For more information, see the sections on International Neuroblastoma Risk Group Staging System (INRGSS) and Children's Oncology Group (COG) Neuroblastoma Risk Grouping.
Treatment era
Between 1975 and 2017, the 5-year survival rate for neuroblastoma in the United States increased from 86% to 91% for children younger than 1 year and from 34% to 83% for children aged 1 to 14 years.[2,3] The 5-year relative survival rate for all infants and children with neuroblastoma increased from 46% when diagnosed between 1974 and 1989 to 71% when diagnosed between 1999 and 2005.[76] More recent estimates from 2011 to 2017 show an even higher survival rate of approximately 85% for infants and children younger than 15 years.[2] These statistics can be misleading because of the extremely heterogeneous prognosis based on the patient's age, stage, and biology. However, studies demonstrate a significant improvement in survival for high-risk patients diagnosed and treated between 2000 and 2010, compared with patients diagnosed from 1990 to 1999.[77] For more information, see Table 1. Similarly, the COG ANBL0531 (NCT00499616) study found equivalent outcomes for many subsets of intermediate-risk children who were treated with substantially reduced chemotherapy, compared with the earlier COG-A3961 (NCT00003093) study.[78]
Age at diagnosis
Infants and children
The effect of age at diagnosis on 5-year survival is profound. In the COG ANBL00B1 (NCT00904241) study of 4,832 patients with newly diagnosed neuroblastoma, those younger than 18 months had a 5-year EFS rate of 82% and an OS rate of 91%. In comparison, patients aged 18 months or older had a 5-year EFS rate of 64% and an OS rate of 74%.[79]
According to the National Childhood Cancer Registry (NCCR), the 5-year relative survival rates from 2011 to 2017 were as follows:[2]
- Aged younger than 1 year: 91%.
- Aged 1 to 4 years: 79%.
- Aged 5 to 9 years: 79%.
- Aged 10 to 14 years: 91%.
The effect of patient age on prognosis is strongly influenced by clinical and pathobiological factors, as evidenced by the following:
- Since 2000, nonrandomized studies of low-risk and intermediate-risk patients have demonstrated that patient age has no effect on outcome of INSS stage 1 or stage 2A disease. However, stage 2B patients younger than 18 months had a 5-year OS rate of 99% (± 1%) versus 90% (± 4%) for children aged 18 months and older.[80]
- In the COG intermediate-risk study A3961 (NCT00003093) that included only MYCN-nonamplified tumors, infants with INSS stage 3 tumors were compared with children with INSS stage 3 favorable-histology tumors. When INSS stage 3 infants with any histology were compared with stage 3 children with favorable histology, only EFS rates, not OS rates, were significantly different (3-year EFS rate, 95% ± 2% vs. 87% ± 3%; OS rate, 98% ± 1% vs. 99% ± 1%).[81]
- Infants younger than 12 months with INSS stage 4 disease and MYCN amplification are categorized as high risk and have a 5-year EFS rate of 37% and an OS rate of 45%.[79] Toddlers aged 12 months to younger than 18 months with stage 4 disease and MYCN-amplified tumors had a 5-year EFS rate of 53% and an OS rate of 54%.[79]
Adolescents and young adults
Adolescents and adults rarely develop neuroblastoma, accounting for less than 5% of all cases. When neuroblastoma occurs in this age range, it shows a more indolent clinical course than neuroblastoma in younger patients, and it shows de novo chemotherapy resistance.[82] Neuroblastoma in adolescents and young adults may also exhibit unusual clinicopathological characteristics such as large tumors, bilateral adrenal disease, and pheochromocytoma-like features.[83][Level of evidence C1] Neuroblastoma has a worse long-term prognosis in adolescents older than 10 years or in adults, regardless of stage or site.
Although adolescent and young adult patients have infrequent MYCN amplification (9% in patients aged 10–21 years), older children with advanced disease have a poor rate of survival. Tumors from the adolescent and young adult population commonly have segmental chromosomal aberrations, and ALK and ATRX variants are much more frequent.[84,85,86] In adolescents, approximately 40% of the tumors have loss-of-function variants in ATRX, compared with less than 20% in younger children and 0% in infants younger than 1 year.[82] Complex DNA microarray findings and novel variants have been reported in some patients.[83][Level of evidence C1]
The 5-year OS rate for adolescent and young adult patients (aged 15–39 years) is 38%.[87][Level of evidence C1] The 5-year EFS rate is 32% for patients between the ages of 10 years and 21 years, and the OS rate is 46%. For patients with stage 4 disease, the 10-year EFS rate is 3%, and the OS rate is 5%.[88] Aggressive chemotherapy and surgery have been shown to achieve a minimal disease state in more than 50% of these patients.[38,89] Other modalities, such as local radiation therapy, autologous stem cell transplant, and the use of agents with confirmed activity, may improve the poor prognosis for adolescents and adults.[88,89]
Adults
The biology of adult-onset neuroblastoma appears to differ from the biology of pediatric or adolescent neuroblastoma based on a single-institution series of 44 patients (aged 18–71 years).[90]
- Genetic abnormalities in adult patients included somatic ATRX (58%) and ALK variants (42%) but no MYCN amplification.
- Germline testing was performed in four patients, two of whom had aberrations (one patient with a BRCA1 pathogenic variant, the other patient with TP53 and NF1 pathogenic variants).
- In the 11 patients with locoregional disease, the 10-year progression-free survival (PFS) rate was 35%, and the OS rate was 61%.
- Among 33 adults with stage 4 neuroblastoma, 7 patients (21%) achieved a complete response (CR) after induction chemotherapy and/or surgery. In patients with stage 4 disease at diagnosis, the 5-year PFS rate was 10%, and most patients who were alive with disease at 5 years died of neuroblastoma over the next 5 years. The 10-year OS rate was 19%. CR after induction was the only prognostic factor for PFS and OS.
- Anti-GD2 immunotherapy (m3F8 or hu3F8) was well tolerated in adults.
As noted above, adult-onset neuroblastoma is enriched for activating ALK variants. In a single-institution retrospective study, 13 adults (median age, 34 years; range, 16–71 years) with relapsed, ALK-altered neuroblastoma were treated with lorlatinib. Nine patients (69%) had a complete or partial response, five of whom were previously treated with other ALK inhibitors. Lorlatinib was associated with significant adverse events requiring dose reduction. However, responses were seen using doses below the recommended adult dose.[91]
Tumor histology
Neuroblastoma tumor histology has a significant impact on prognosis and risk group assignment. For more information, see the Classification of Neuroblastic Tumors section and Table 3.
In the ANBL00B1 (NCT00904241) study of 4,832 patients with newly diagnosed neuroblastoma, 52% of patients were classified as favorable and 48% as unfavorable, according to the International Neuroblastoma Pathology Classification (INPC). For patients with tumors classified as favorable, the 5-year EFS rate was 88%, and the 5-year OS rate was 96%. For patients with tumors classified as unfavorable, the 5-year EFS rate was 55%, and the 5-year OS rate was 66% (P < .0001).[79]
Histological characteristics considered prognostically favorable include the following:
- Cellular differentiation/maturation. Higher degrees of neuroblastic maturation confer improved prognosis for stage 4 patients with segmental chromosome changes without MYCN amplification. Neuroblastoma tumors containing many differentiating cells, termed ganglioneuroblastoma, can have diffuse differentiation conferring a very favorable prognosis or can have nodules of undifferentiated cells, termed nodular ganglioneuroblastoma, whose histology, along with MYCN status, determine prognosis.[92,93]
- Schwannian stroma.
- Cystic neuroblastoma. About 25% of reported neuroblastomas diagnosed in the fetus and neonate are cystic. Patients with cystic neuroblastomas have tumors with lower disease stages and a higher incidence of favorable biology.[94]
High mitosis/karyorrhexis index and undifferentiated tumor cells are considered prognostically unfavorable histological characteristics, but the prognostic value is age dependent.[95,96]
A COG study (P9641 [NCT00003119]) investigated the effect of histology, among other factors, on outcome. Of 915 children with stage 1 and stage 2 neuroblastoma without MYCN amplification, 87% were treated with initial surgery and observation. Patients (13%) who had or were at risk of developing symptomatic disease, or who had less than 50% tumor resection at diagnosis, or who had unresectable progressive disease after surgery alone, were treated with chemotherapy and surgery. Those with favorable histological features reported a 5-year EFS rate of 90% to 94% and an OS rate of 99% to 100%. Those with unfavorable histology had an EFS rate of 80% to 86% and an OS rate of 89% to 93%.[80]
- In the COG ANBL0531 (NCT00499616) study for intermediate-risk patients with neuroblastoma, treatment was assigned using a biology-based and response-based algorithm, which included allelic status of 1p36 and 11q23. Patients with MYCN-amplified tumors were excluded.[78]
- EFS was statistically significantly better for infants with stage 4 disease with favorable tumor biology (n = 61) (3-year EFS rate, 86.9%; 95% CI, 78.3%–95.4%), compared with those with confirmed unfavorable tumor biology (n = 47) (3-year EFS rate, 66.8%; 95% CI, 53.1%–80.6%; P = .02). With longer follow-up, the 10-year EFS rates were 86.9% for infants with stage 4 tumors that had favorable biology versus 66.8% (P = .02) for infants with tumors that had unfavorable biology.[97]
- OS for infants with stage 4 disease and favorable tumor biology showed a trend toward OS advantage (3-year OS rate, 95.0%; 95% CI, 89.5%–100% vs. 86.7%; 95% CI, 76.6%–96.7%; P = .08). However, with longer follow-up, the 10-year OS rates were not significantly different between infants with stage 4 tumors that had favorable biology and those with tumors that had unfavorable biology (95.0% vs. 84.4%; P = .08).[97]
- Among the group 4 infants (n = 24) with stage 4 disease with confirmed diploid or unfavorable histology tumors, with or without 1p36/11q23 loss of heterozygosity, the 3-year EFS rate estimate was 63.9% (95% CI, 43.8%–84.0%), and the 3-year OS rate estimate was 77.3% (95% CI, 59.2%–95.3%).
- For infants with stage 4 hyperdiploid favorable histology tumors assigned to group 4 because of 1p36/11q23 loss of heterozygosity or unknown allelic status (n = 32), the 3-year EFS and OS rate estimates were 68.6% (95% CI, 52.2%–85.1%) and 93.8% (95% CI, 85.2%–100%), respectively.
- The EFS and OS rate estimates for the eight toddlers (aged 12–18 months) with stage 4 hyperdiploid favorable histology tumors were 62.5% (95% CI, 28.9%–96.1%) and 100%, respectively.
- Patients with favorable biology and localized disease had a 100% survival rate.
A study using data from the INRG Data Commons evaluated the prognostic strength of the underlying INPC histological criteria. The independent prognostic ability of age, histological category, mitosis-karyorrhexis index (MKI), and grade was demonstrated. Four age-related, histological prognostic groups were identified (aged <18 months with low vs. high MKI, and aged ≥18 months with differentiated vs. undifferentiated/poorly differentiated tumors). Compared with survival trees generated with established COG risk criteria, an additional prognostic subgroup was identified and validated when individual histological features were analyzed in lieu of INPC. Thus, replacing INPC with individual histological features in the future COG risk classification may eliminate the duplication of the prognostic contribution of age, facilitate international harmonization of risk classification, and provide a schema for more precise prognostication and refined therapeutic approaches.[98] The INPC is described in the Classification of Neuroblastic Tumors section.
Biological features
For more information, see the Genomic and Biological Features of Neuroblastoma section.
Site of primary tumor
Clinical and biological features of neuroblastoma differ by primary tumor site. In a study of data on 8,389 patients in clinical trials and compiled by the International Risk Group Project, the following results were observed, confirming the results from much smaller, previous studies with less complete clinical and biological data:[99]
- Adrenal tumors. Adrenal primary tumors were more likely than tumors originating in other sites to be associated with unfavorable prognostic features, including MYCN amplification, even after researchers controlled for age, stage, and histological grade. Adrenal neuroblastomas were also associated with a higher incidence of stage 4 tumors, segmental chromosomal aberrations, diploidy, unfavorable INPC histology, age younger than 18 months, and elevated levels of LDH and ferritin. The relative risk of MYCN amplification compared with adrenal tumors was 0.7 in abdominal nonadrenal tumors and about 0.1 in nonabdominal paraspinal tumors.
- Thoracic tumors. Thoracic tumors were compared with nonthoracic tumors. After researchers controlled for age, stage, and histological grade, results showed patients with thoracic tumors had fewer deaths and recurrences (HR, 0.79; 95% CI, 0.67–0.92), and thoracic tumors had a lower incidence of MYCN amplification (adjusted OR, 0.20; 95% CI, 0.11–0.39).
Using the Therapeutically Applicable Research to Generate Effect Treatments (TARGET) and genome-wide association study data sets, a study compared the genomic and epigenomic data of primary diagnostic neuroblastomas originating in the adrenal gland (n = 646) with that of neuroblastomas originating in the thoracic sympathetic ganglia (n = 118). Neuroblastomas arising in the adrenal gland were more likely to harbor structural DNA aberrations such as MYCN amplification, whereas thoracic tumors showed defects in mitotic checkpoints resulting in hyperdiploidy. Thoracic tumors were more likely to harbor gain-of-function ALK aberrations than were adrenal tumors among all cases (OR, 1.89; P = .04), and among cases without MYCN amplification (OR, 2.86; P = .003). Because 16% of thoracic tumors harbor ALK variants, routine sequencing for these variants in this setting should be considered.[100]
In the TARGET cohort, 70% of patients with adrenal primary tumors and 51% of patients with thoracic primary tumors were stage 4. In the genome-wide association study without MYCN amplification, 43% of patients with adrenal primary tumors and 17% of patients with thoracic primary tumors were stage 4. By multivariate analysis, adrenal site was an independent predictor of worse outcome in the genome-wide association study cohort but not in the TARGET cohort after adjusting for MYCN amplification status, disease stage, and age of at least 18 months. Adrenal neuroblastoma was not an independent predictor of worse EFS by similar multivariable analysis for either the genome-wide association study or TARGET cohorts.[100]
It is not clear whether the effect of primary neuroblastoma tumor site on prognosis is entirely dependent on the differences in tumor biology associated with tumor site.
Multifocal neuroblastoma occurs rarely, usually in infants, and generally has a good prognosis.[101] Familial neuroblastoma and germline ALK gene pathogenic variants should be considered in patients with multiple primary neuroblastomas.
Stage of disease
Several imaged-based and surgery-based systems were used for assigning disease stage before the 1990s. In an effort to compare results obtained throughout the world, a surgical pathological staging system, termed the International Neuroblastoma Staging System (INSS), was developed.[62] The INSS predicted outcome on the basis of stage at diagnosis, although important interactions with biological variables were also found.[3,4,11,62,80,81,102,103,104] However, because surgical approaches differ from one institution to another, INSS stage for patients with locoregional disease may also vary considerably. To define extent of disease at diagnosis in a uniform manner, a presurgical International Neuroblastoma Risk Group staging system (INRGSS) was developed for the International Neuroblastoma Risk Group Classification System.[105,106] The INRGSS is currently used in North American and European cooperative group studies. This staging system is not affected by locoregional lymph node involvement.
For the patients with newly diagnosed neuroblastoma enrolled in the ANBL00B1 (NCT00904241) study, the 5-years EFS and OS rates, according to INRGSS stage, were the following:[79]
- 90% and 98% for patients with stage L1 disease.
- 84% and 95% for patients with stage L2 disease.
- 52% and 64% for patients with stage M disease.
- 86% and 92% for patients with stage MS disease.
For more information, see the following sections:
- International Neuroblastoma Staging System (INSS).
- International Neuroblastoma Risk Group Staging System (INRGSS).
- Treatment Options for Low-Risk Neuroblastoma, Evidence (for removal of chemotherapy).
- Treatment Options for Intermediate-Risk Neuroblastoma, Evidence (chemotherapy with or without surgery).
- Treatment Options for Intermediate-Risk Neuroblastoma, Radiation therapy.
- Treatment Options for High-Risk Neuroblastoma.
- Treatment Options for Stage 4S/MS Neuroblastoma.
Response to treatment
Response to treatment has been associated with outcome. In patients with intermediate-risk disease who had a poor response to initial therapy in the COG ANBL0531 (NCT00499616) study, 6 of 20 patients subsequently developed progressive or recurrent disease, and one patient died.[78]
In patients with high-risk disease, the persistence of neuroblastoma cells in bone marrow after induction chemotherapy, for example, is associated with a poor prognosis. Sensitive techniques that detect minimal residual disease may be used to assess prognosis.[107,108,109] Similarly, the persistence of MIBG-avid tumor measured as Curie score greater than 2 after completion of induction therapy predicts a poor prognosis for patients with MYCN-nonamplified high-risk tumors. A Curie score greater than 0 after induction therapy is associated with a worse outcome for high-risk patients with MYCN-amplified disease.[110,111] An analysis of North American patients who went on to receive tandem transplants showed that patients with Curie scores greater than 0 at the end of induction therapy had inferior EFS rates.[112] For more information about Curie scoring, see the Curie and SIOPEN scoring methods section.
In an analysis of patients from four consecutive COG high-risk trials, an end-induction response of partial response (PR) or better, according to the 1993 International Neuroblastoma Response Criteria,[62] was significantly associated with higher EFS and OS. On multivariable analysis (n = 407), the absence of 11q loss of heterozygosity (LOH) was the only factor that remained significantly associated with PR or better (OR, 1.962 vs. 11q LOH; 95% CI, 1.104–3.487; P = .0216).[113]
A treatment-associated decrease in mitosis and an increase in histological differentiation of the primary tumor are also prognostic of response.[114]
The accuracy of prognostication based on decrease in primary tumor size is less clear. In a study conducted by seven large international centers, 229 high-risk patients were treated in a variety of ways. Treatment included chemotherapy, surgical removal of the primary tumor, radiation to the tumor bed, high-dose myeloablative therapy plus stem cell transplant, and, in most cases, isotretinoin and anti-GD2 antibody immunotherapy enhanced by cytokines. Primary tumor response was measured after induction chemotherapy in three ways: as 30% or greater reduction in the longest dimension, 50% or greater reduction in tumor volume, or 65% or greater reduction in tumor volume (calculated from three tumor dimensions, a conventional radiological technique). The measurements were performed at diagnosis and after induction chemotherapy before primary tumor resection. None of the methods of measuring primary tumor response at end of induction chemotherapy predicted survival.[115]
Levels of LDH and ferritin
Higher serum LDH and ferritin values conferred worse 5-year EFS and OS rates in a large international cohort of patients diagnosed with neuroblastoma (n > 8,575) from 1990 to 2016. Higher serum values for LDH and ferritin also conferred worse 3-year EFS and OS rates in patients with high-risk neuroblastoma after 2009. In a multivariate analysis that adjusted for age at diagnosis, MYCN status, and INSS stage 4 disease, LDH and ferritin maintained independent prognostic ability (P < .0001).[116][Level of evidence C1]
Although not critically evaluated in the original INRG classification system, subsequent analysis of the INRG Data Commons has clearly demonstrated independent statistical significance of the levels of serum ferritin and LDH on prognosis in all patients and in high-risk patients, including in the time period between 2010 and 2016. Therefore, it was suggested that these two easily obtainable lab values be incorporated into the prognostic classification system of the INRG.[116]
References:
- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 29. Also available online. Last accessed August 21, 2023.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Childhood cancer. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 28. Also available online. Last accessed August 21, 2023.
- Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 30, 2024.
- Gurney JG, Ross JA, Wall DA, et al.: Infant cancer in the U.S.: histology-specific incidence and trends, 1973 to 1992. J Pediatr Hematol Oncol 19 (5): 428-32, 1997 Sep-Oct.
- United States Census Bureau: Age and Sex Composition in the United States: 2018. U.S. Census Bureau, 2018. Available online. Last accessed August 21, 2023.
- Mahapatra S, Challagundla KB: Neuroblastoma. Treasure Island, FL: StatPearls Publishing LLC, 2022. Available online. Last accessed August 21, 2023.
- Campbell K, Siegel DA, Umaretiya PJ, et al.: A comprehensive analysis of neuroblastoma incidence, survival, and racial and ethnic disparities from 2001 to 2019. Pediatr Blood Cancer 71 (1): e30732, 2024.
- London WB, Castleberry RP, Matthay KK, et al.: Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children's Oncology Group. J Clin Oncol 23 (27): 6459-65, 2005.
- Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations). National Cancer Institute, 2012. Also available online. Last accessed May 22, 2024.
- Takeuchi LA, Hachitanda Y, Woods WG, et al.: Screening for neuroblastoma in North America. Preliminary results of a pathology review from the Quebec Project. Cancer 76 (11): 2363-71, 1995.
- Woods WG, Gao RN, Shuster JJ, et al.: Screening of infants and mortality due to neuroblastoma. N Engl J Med 346 (14): 1041-6, 2002.
- Schilling FH, Spix C, Berthold F, et al.: Neuroblastoma screening at one year of age. N Engl J Med 346 (14): 1047-53, 2002.
- Heck JE, Ritz B, Hung RJ, et al.: The epidemiology of neuroblastoma: a review. Paediatr Perinat Epidemiol 23 (2): 125-43, 2009.
- Mossé YP, Laudenslager M, Longo L, et al.: Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455 (7215): 930-5, 2008.
- Mosse YP, Laudenslager M, Khazi D, et al.: Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet 75 (4): 727-30, 2004.
- Raabe EH, Laudenslager M, Winter C, et al.: Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene 27 (4): 469-76, 2008.
- van Limpt V, Schramm A, van Lakeman A, et al.: The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene 23 (57): 9280-8, 2004.
- Serra A, Häberle B, König IR, et al.: Rare occurrence of PHOX2b mutations in sporadic neuroblastomas. J Pediatr Hematol Oncol 30 (10): 728-32, 2008.
- Satgé D, Moore SW, Stiller CA, et al.: Abnormal constitutional karyotypes in patients with neuroblastoma: a report of four new cases and review of 47 others in the literature. Cancer Genet Cytogenet 147 (2): 89-98, 2003.
- Mosse Y, Greshock J, King A, et al.: Identification and high-resolution mapping of a constitutional 11q deletion in an infant with multifocal neuroblastoma. Lancet Oncol 4 (12): 769-71, 2003.
- Moroni I, Bedeschi F, Luksch R, et al.: Costello syndrome: a cancer predisposing syndrome? Clin Dysmorphol 9 (4): 265-8, 2000.
- Cotton JL, Williams RG: Noonan syndrome and neuroblastoma. Arch Pediatr Adolesc Med 149 (11): 1280-1, 1995.
- Gutmann DH, Ferner RE, Listernick RH, et al.: Neurofibromatosis type 1. Nat Rev Dis Primers 3: 17004, 2017.
- Kamihara J, Bourdeaut F, Foulkes WD, et al.: Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clin Cancer Res 23 (13): e98-e106, 2017.
- Bougnères P, Pantalone L, Linglart A, et al.: Endocrine manifestations of the rapid-onset obesity with hypoventilation, hypothalamic, autonomic dysregulation, and neural tumor syndrome in childhood. J Clin Endocrinol Metab 93 (10): 3971-80, 2008.
- Maas SM, Vansenne F, Kadouch DJ, et al.: Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A 170 (9): 2248-60, 2016.
- Witkowski L, Nichols KE, Jongmans M, et al.: Germline pathogenic SMARCA4 variants in neuroblastoma. J Med Genet 60 (10): 987-992, 2023.
- Kim J, Vaksman Z, Egolf LE, et al.: Germline pathogenic variants in neuroblastoma patients are enriched in BARD1 and predict worse survival. J Natl Cancer Inst 116 (1): 149-159, 2024.
- Tolbert VP, Coggins GE, Maris JM: Genetic susceptibility to neuroblastoma. Curr Opin Genet Dev 42: 81-90, 2017.
- Matser YAH, Verly IRN, van der Ham M, et al.: Optimising urinary catecholamine metabolite diagnostics for neuroblastoma. Pediatr Blood Cancer 70 (6): e30289, 2023.
- Kratz CP, Rapisuwon S, Reed H, et al.: Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet 157 (2): 83-9, 2011.
- Mussa A, Molinatto C, Baldassarre G, et al.: Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J Pediatr 176: 142-149.e1, 2016.
- Citak C, Karadeniz C, Dalgic B, et al.: Intestinal lymphangiectasia as a first manifestation of neuroblastoma. Pediatr Blood Cancer 46 (1): 105-7, 2006.
- Bourdeaut F, de Carli E, Timsit S, et al.: VIP hypersecretion as primary or secondary syndrome in neuroblastoma: A retrospective study by the Société Française des Cancers de l'Enfant (SFCE). Pediatr Blood Cancer 52 (5): 585-90, 2009.
- Mahoney NR, Liu GT, Menacker SJ, et al.: Pediatric horner syndrome: etiologies and roles of imaging and urine studies to detect neuroblastoma and other responsible mass lesions. Am J Ophthalmol 142 (4): 651-9, 2006.
- Conte M, Parodi S, De Bernardi B, et al.: Neuroblastoma in adolescents: the Italian experience. Cancer 106 (6): 1409-17, 2006.
- Matthay KK, Blaes F, Hero B, et al.: Opsoclonus myoclonus syndrome in neuroblastoma a report from a workshop on the dancing eyes syndrome at the advances in neuroblastoma meeting in Genoa, Italy, 2004. Cancer Lett 228 (1-2): 275-82, 2005.
- Rudnick E, Khakoo Y, Antunes NL, et al.: Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: clinical outcome and antineuronal antibodies-a report from the Children's Cancer Group Study. Med Pediatr Oncol 36 (6): 612-22, 2001.
- Antunes NL, Khakoo Y, Matthay KK, et al.: Antineuronal antibodies in patients with neuroblastoma and paraneoplastic opsoclonus-myoclonus. J Pediatr Hematol Oncol 22 (4): 315-20, 2000 Jul-Aug.
- Pang KK, de Sousa C, Lang B, et al.: A prospective study of the presentation and management of dancing eye syndrome/opsoclonus-myoclonus syndrome in the United Kingdom. Eur J Paediatr Neurol 14 (2): 156-61, 2010.
- Pranzatelli MR: The neurobiology of the opsoclonus-myoclonus syndrome. Clin Neuropharmacol 15 (3): 186-228, 1992.
- Mitchell WG, Davalos-Gonzalez Y, Brumm VL, et al.: Opsoclonus-ataxia caused by childhood neuroblastoma: developmental and neurologic sequelae. Pediatrics 109 (1): 86-98, 2002.
- Cooper R, Khakoo Y, Matthay KK, et al.: Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: histopathologic features-a report from the Children's Cancer Group. Med Pediatr Oncol 36 (6): 623-9, 2001.
- Pranzatelli MR, Tate ED, McGee NR: Demographic, Clinical, and Immunologic Features of 389 Children with Opsoclonus-Myoclonus Syndrome: A Cross-sectional Study. Front Neurol 8: 468, 2017.
- Hero B, Clement N, Øra I, et al.: Genomic Profiles of Neuroblastoma Associated With Opsoclonus Myoclonus Syndrome. J Pediatr Hematol Oncol 40 (2): 93-98, 2018.
- Catsman-Berrevoets CE, Aarsen FK, van Hemsbergen ML, et al.: Improvement of neurological status and quality of life in children with opsoclonus myoclonus syndrome at long-term follow-up. Pediatr Blood Cancer 53 (6): 1048-53, 2009.
- Connolly AM, Pestronk A, Mehta S, et al.: Serum autoantibodies in childhood opsoclonus-myoclonus syndrome: an analysis of antigenic targets in neural tissues. J Pediatr 130 (6): 878-84, 1997.
- Bell J, Moran C, Blatt J: Response to rituximab in a child with neuroblastoma and opsoclonus-myoclonus. Pediatr Blood Cancer 50 (2): 370-1, 2008.
- Corapcioglu F, Mutlu H, Kara B, et al.: Response to rituximab and prednisolone for opsoclonus-myoclonus-ataxia syndrome in a child with ganglioneuroblastoma. Pediatr Hematol Oncol 25 (8): 756-61, 2008.
- Pranzatelli MR, Tate ED, Travelstead AL, et al.: Rituximab (anti-CD20) adjunctive therapy for opsoclonus-myoclonus syndrome. J Pediatr Hematol Oncol 28 (9): 585-93, 2006.
- Ertle F, Behnisch W, Al Mulla NA, et al.: Treatment of neuroblastoma-related opsoclonus-myoclonus-ataxia syndrome with high-dose dexamethasone pulses. Pediatr Blood Cancer 50 (3): 683-7, 2008.
- Pranzatelli MR, Tate ED: Dexamethasone, Intravenous Immunoglobulin, and Rituximab Combination Immunotherapy for Pediatric Opsoclonus-Myoclonus Syndrome. Pediatr Neurol 73: 48-56, 2017.
- de Alarcon PA, Matthay KK, London WB, et al.: Intravenous immunoglobulin with prednisone and risk-adapted chemotherapy for children with opsoclonus myoclonus ataxia syndrome associated with neuroblastoma (ANBL00P3): a randomised, open-label, phase 3 trial. Lancet Child Adolesc Health 2 (1): 25-34, 2018.
- Kumar P, Willard VW, Embry L, et al.: Late cognitive and adaptive outcomes of patients with neuroblastoma-associated opsoclonus-myoclonus-ataxia-syndrome: A report from the Children's Oncology Group. Pediatr Blood Cancer 71 (7): e31039, 2024.
- Vik TA, Pfluger T, Kadota R, et al.: (123)I-mIBG scintigraphy in patients with known or suspected neuroblastoma: Results from a prospective multicenter trial. Pediatr Blood Cancer 52 (7): 784-90, 2009.
- Yang J, Codreanu I, Servaes S, et al.: I-131 MIBG post-therapy scan is more sensitive than I-123 MIBG pretherapy scan in the evaluation of metastatic neuroblastoma. Nucl Med Commun 33 (11): 1134-7, 2012.
- Sharp SE, Shulkin BL, Gelfand MJ, et al.: 123I-MIBG scintigraphy and 18F-FDG PET in neuroblastoma. J Nucl Med 50 (8): 1237-43, 2009.
- Pio L, Brisse HJ, Alaggio R, et al.: Image-guided core-needle or surgical biopsy for neuroblastoma diagnosis in children: A systematic review and meta-analysis from the International Society of Pediatric Surgical Oncology (IPSO). Pediatr Blood Cancer 71 (2): e30789, 2024.
- Schoeman S, Bagatell R, Cahill AM, et al.: Percutaneous biopsy for the diagnosis, risk stratification, and molecular profiling of neuroblastoma: A single-center retrospective study. Pediatr Blood Cancer 71 (4): e30887, 2024.
- Brodeur GM, Pritchard J, Berthold F, et al.: Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11 (8): 1466-77, 1993.
- Nickerson HJ, Matthay KK, Seeger RC, et al.: Favorable biology and outcome of stage IV-S neuroblastoma with supportive care or minimal therapy: a Children's Cancer Group study. J Clin Oncol 18 (3): 477-86, 2000.
- Jennings RW, LaQuaglia MP, Leong K, et al.: Fetal neuroblastoma: prenatal diagnosis and natural history. J Pediatr Surg 28 (9): 1168-74, 1993.
- Brodeur GM: Spontaneous regression of neuroblastoma. Cell Tissue Res 372 (2): 277-286, 2018.
- Guan J, Hallberg B, Palmer RH: Chromosome Imbalances in Neuroblastoma-Recent Molecular Insight into Chromosome 1p-deletion, 2p-gain, and 11q-deletion Identifies New Friends and Foes for the Future. Cancers (Basel) 13 (23): , 2021.
- Schneiderman J, London WB, Brodeur GM, et al.: Clinical significance of MYCN amplification and ploidy in favorable-stage neuroblastoma: a report from the Children's Oncology Group. J Clin Oncol 26 (6): 913-8, 2008.
- Hiyama E, Hiyama K, Yokoyama T, et al.: Correlating telomerase activity levels with human neuroblastoma outcomes. Nat Med 1 (3): 249-55, 1995.
- Kitanaka C, Kato K, Ijiri R, et al.: Increased Ras expression and caspase-independent neuroblastoma cell death: possible mechanism of spontaneous neuroblastoma regression. J Natl Cancer Inst 94 (5): 358-68, 2002.
- Brodeur GM, Minturn JE, Ho R, et al.: Trk receptor expression and inhibition in neuroblastomas. Clin Cancer Res 15 (10): 3244-50, 2009.
- Yamamoto K, Ohta S, Ito E, et al.: Marginal decrease in mortality and marked increase in incidence as a result of neuroblastoma screening at 6 months of age: cohort study in seven prefectures in Japan. J Clin Oncol 20 (5): 1209-14, 2002.
- Okazaki T, Kohno S, Mimaya J, et al.: Neuroblastoma detected by mass screening: the Tumor Board's role in its treatment. Pediatr Surg Int 20 (1): 27-32, 2004.
- Fritsch P, Kerbl R, Lackner H, et al.: "Wait and see" strategy in localized neuroblastoma in infants: an option not only for cases detected by mass screening. Pediatr Blood Cancer 43 (6): 679-82, 2004.
- Nuchtern JG, London WB, Barnewolt CE, et al.: A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: a Children's Oncology Group study. Ann Surg 256 (4): 573-80, 2012.
- Hero B, Simon T, Spitz R, et al.: Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. J Clin Oncol 26 (9): 1504-10, 2008.
- Horner MJ, Ries LA, Krapcho M, et al.: SEER Cancer Statistics Review, 1975-2006. National Cancer Institute, 2009. Also available online. Last accessed August 21, 2023.
- Pinto NR, Applebaum MA, Volchenboum SL, et al.: Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J Clin Oncol 33 (27): 3008-17, 2015.
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.
- Strother DR, London WB, Schmidt ML, et al.: Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30 (15): 1842-8, 2012.
- Baker DL, Schmidt ML, Cohn SL, et al.: Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med 363 (14): 1313-23, 2010.
- Cheung NK, Zhang J, Lu C, et al.: Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 307 (10): 1062-71, 2012.
- McCarthy LC, Chastain K, Flatt TG, et al.: Neuroblastoma in Adolescents and Children Older than 10 Years: Unusual Clinicopathologic and Biologic Features. J Pediatr Hematol Oncol 41 (8): 586-595, 2019.
- Mazzocco K, Defferrari R, Sementa AR, et al.: Genetic abnormalities in adolescents and young adults with neuroblastoma: A report from the Italian Neuroblastoma group. Pediatr Blood Cancer 62 (10): 1725-32, 2015.
- Defferrari R, Mazzocco K, Ambros IM, et al.: Influence of segmental chromosome abnormalities on survival in children over the age of 12 months with unresectable localised peripheral neuroblastic tumours without MYCN amplification. Br J Cancer 112 (2): 290-5, 2015.
- Pugh TJ, Morozova O, Attiyeh EF, et al.: The genetic landscape of high-risk neuroblastoma. Nat Genet 45 (3): 279-84, 2013.
- Chen I, Pasalic D, Fischer-Valuck B, et al.: Disparity in Outcomes for Adolescent and Young Adult Patients Diagnosed With Pediatric Solid Tumors Across 4 Decades. Am J Clin Oncol 41 (5): 471-475, 2018.
- Mossé YP, Deyell RJ, Berthold F, et al.: Neuroblastoma in older children, adolescents and young adults: a report from the International Neuroblastoma Risk Group project. Pediatr Blood Cancer 61 (4): 627-35, 2014.
- Kushner BH, Kramer K, LaQuaglia MP, et al.: Neuroblastoma in adolescents and adults: the Memorial Sloan-Kettering experience. Med Pediatr Oncol 41 (6): 508-15, 2003.
- Suzuki M, Kushner BH, Kramer K, et al.: Treatment and outcome of adult-onset neuroblastoma. Int J Cancer 143 (5): 1249-1258, 2018.
- Stiefel J, Kushner BH, Roberts SS, et al.: Anaplastic Lymphoma Kinase Inhibitors for Therapy of Neuroblastoma in Adults. JCO Precis Oncol 7: e2300138, 2023.
- Kubota M, Suita S, Tajiri T, et al.: Analysis of the prognostic factors relating to better clinical outcome in ganglioneuroblastoma. J Pediatr Surg 35 (1): 92-5, 2000.
- Peuchmaur M, d'Amore ES, Joshi VV, et al.: Revision of the International Neuroblastoma Pathology Classification: confirmation of favorable and unfavorable prognostic subsets in ganglioneuroblastoma, nodular. Cancer 98 (10): 2274-81, 2003.
- Isaacs H: Fetal and neonatal neuroblastoma: retrospective review of 271 cases. Fetal Pediatr Pathol 26 (4): 177-84, 2007 Jul-Aug.
- Ikeda H, Iehara T, Tsuchida Y, et al.: Experience with International Neuroblastoma Staging System and Pathology Classification. Br J Cancer 86 (7): 1110-6, 2002.
- Teshiba R, Kawano S, Wang LL, et al.: Age-dependent prognostic effect by Mitosis-Karyorrhexis Index in neuroblastoma: a report from the Children's Oncology Group. Pediatr Dev Pathol 17 (6): 441-9, 2014 Nov-Dec.
- Barr EK, Naranjo A, Twist CJ, et al.: Long-term follow-up of patients with intermediate-risk neuroblastoma treated with response- and biology-based therapy: A report from the Children's Oncology Group study ANBL0531. Pediatr Blood Cancer 71 (8): e31089, 2024.
- Sokol E, Desai AV, Applebaum MA, et al.: Age, Diagnostic Category, Tumor Grade, and Mitosis-Karyorrhexis Index Are Independently Prognostic in Neuroblastoma: An INRG Project. J Clin Oncol 38 (17): 1906-1918, 2020.
- Vo KT, Matthay KK, Neuhaus J, et al.: Clinical, biologic, and prognostic differences on the basis of primary tumor site in neuroblastoma: a report from the international neuroblastoma risk group project. J Clin Oncol 32 (28): 3169-76, 2014.
- Oldridge DA, Truong B, Russ D, et al.: Differences in Genomic Profiles and Outcomes Between Thoracic and Adrenal Neuroblastoma. J Natl Cancer Inst 111 (11): 1192-1201, 2019.
- Hiyama E, Yokoyama T, Hiyama K, et al.: Multifocal neuroblastoma: biologic behavior and surgical aspects. Cancer 88 (8): 1955-63, 2000.
- Ward E, DeSantis C, Robbins A, et al.: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 64 (2): 83-103, 2014 Mar-Apr.
- Bagatell R, Beck-Popovic M, London WB, et al.: Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol 27 (3): 365-70, 2009.
- Campbell K, Gastier-Foster JM, Mann M, et al.: Association of MYCN copy number with clinical features, tumor biology, and outcomes in neuroblastoma: A report from the Children's Oncology Group. Cancer 123 (21): 4224-4235, 2017.
- Cohn SL, Pearson AD, London WB, et al.: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27 (2): 289-97, 2009.
- Monclair T, Brodeur GM, Ambros PF, et al.: The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 27 (2): 298-303, 2009.
- Burchill SA, Lewis IJ, Abrams KR, et al.: Circulating neuroblastoma cells detected by reverse transcriptase polymerase chain reaction for tyrosine hydroxylase mRNA are an independent poor prognostic indicator in stage 4 neuroblastoma in children over 1 year. J Clin Oncol 19 (6): 1795-801, 2001.
- Seeger RC, Reynolds CP, Gallego R, et al.: Quantitative tumor cell content of bone marrow and blood as a predictor of outcome in stage IV neuroblastoma: a Children's Cancer Group Study. J Clin Oncol 18 (24): 4067-76, 2000.
- Bochennek K, Esser R, Lehrnbecher T, et al.: Impact of minimal residual disease detection prior to autologous stem cell transplantation for post-transplant outcome in high risk neuroblastoma. Klin Padiatr 224 (3): 139-42, 2012.
- Yanik GA, Parisi MT, Shulkin BL, et al.: Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: a report from the Children's oncology group. J Nucl Med 54 (4): 541-8, 2013.
- Yanik GA, Parisi MT, Naranjo A, et al.: Validation of Postinduction Curie Scores in High-Risk Neuroblastoma: A Children's Oncology Group and SIOPEN Group Report on SIOPEN/HR-NBL1. J Nucl Med 59 (3): 502-508, 2018.
- Streby KA, Parisi MT, Shulkin BL, et al.: Impact of diagnostic and end-of-induction Curie scores with tandem high-dose chemotherapy and autologous transplants for metastatic high-risk neuroblastoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 70 (8): e30418, 2023.
- Pinto N, Naranjo A, Hibbitts E, et al.: Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children's Oncology Group (COG). Eur J Cancer 112: 66-79, 2019.
- George RE, Perez-Atayde AR, Yao X, et al.: Tumor histology during induction therapy in patients with high-risk neuroblastoma. Pediatr Blood Cancer 59 (3): 506-10, 2012.
- Bagatell R, McHugh K, Naranjo A, et al.: Assessment of Primary Site Response in Children With High-Risk Neuroblastoma: An International Multicenter Study. J Clin Oncol 34 (7): 740-6, 2016.
- Moroz V, Machin D, Hero B, et al.: The prognostic strength of serum LDH and serum ferritin in children with neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 67 (8): e28359, 2020.