




General Information About Chronic Lymphocytic Leukemia (CLL)
Incidence and Mortality
Estimated new cases and deaths from CLL in the United States in 2024:[1]
- New cases: 20,700.
- Deaths: 4,440.
Anatomy
CLL is a disorder of morphologically mature, but immunologically less mature lymphocytes. It is manifested by progressive accumulation of these cells in the blood, bone marrow, and lymphatic tissues.[2]
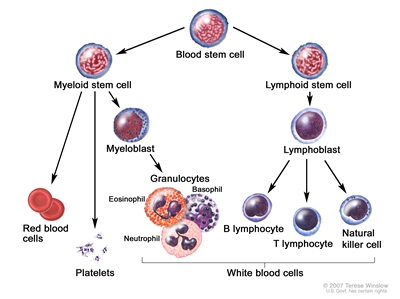
Blood cell development. A blood stem cell goes through several steps to become a red blood cell, platelet, or white blood cell.
Clinical Presentation
The clinical course of this disease progresses from an indolent lymphocytosis without other evident disease to one of generalized lymphatic enlargement with concomitant pancytopenia. Complications of pancytopenia, including hemorrhage and infection, represent a major cause of death in these patients.[3] Immunological aberrations, including Coombs-positive hemolytic anemia, immune thrombocytopenia, and depressed immunoglobulin levels may all complicate the management of CLL.[4]
Diagnostic Evaluation and Differential Diagnosis
Tests and procedures used to diagnose CLL include the following:[5]
- History and physical examination (including bidimensional diameters of the largest palpable lymph nodes in the cervical, axillary, and inguinal nodal sites and dimensions of the liver and spleen below their respective costal margins as assessed by palpation).
- Complete blood count with differential and chemistry panel (including creatinine, bilirubin, transaminases, and alkaline phosphatase). Other blood tests may include lactate dehydrogenase and beta-2-microglobulin. With suspicion of autoimmune hemolytic anemia, testing for reticulocyte count, indirect bilirubin, serum haptoglobin, antiglobulin (direct Coombs), and cold agglutinin may be helpful.
- Flow cytometry (for immunophenotyping).
- Fluorescence in situ hybridization (FISH) (for del(11q), del(13q), del(17p), trisomy 12, and t(11;14)).
- TP53 variant analysis.
- IGH variant analysis.
- Serum immunoglobulin levels.
- Hepatitis B and C and HIV tests.
- Computed tomography (CT) is usually not required in the absence of peripheral adenopathy; extensive adenopathy on examination should prompt investigation of retroperitoneal adenopathy.
- Bone marrow aspiration and biopsy is usually not required.
In this disorder, lymphocyte counts in the blood are usually greater than or equal to 5,000/mm3 with a characteristic immunophenotype (CD5- and CD23-positive B cells).[6,7] As assays have become more sensitive for detecting monoclonal B-CLL–like cells in peripheral blood, researchers have detected a monoclonal B-cell lymphocytosis in 3% of adults older than 40 years and in 6% of adults older than 60 years.[8] Such early detection and diagnosis may falsely suggest improved survival for the group and may unnecessarily worry or result in therapy for some patients who would have remained undiagnosed in their lifetime, a circumstance known as overdiagnosis or pseudodisease.[9,10]
Confusion with other diseases may be avoided by determination of cell surface markers. CLL lymphocytes coexpress the B-cell antigens CD19 and CD20 along with the T-cell antigen CD5.[11] This coexpression occurs in only one other disease entity, mantle cell lymphoma. CLL B cells express relatively low levels of surface-membrane immunoglobulin (compared with normal peripheral blood B cells) and a single light chain (kappa or lambda).[12] CLL is diagnosed by an absolute increase in lymphocytosis and/or bone marrow infiltration coupled with the characteristic features of morphology and immunophenotype, which confirm the characteristic clonal population. In a database analysis, for up to 77 months before diagnosis, almost all patients with a CLL diagnosis had prediagnostic B-cell clones that were identified in peripheral blood (when available).[7,13]
About 1% of morphological CLL cases express T-cell markers (CD4 and CD7) and have clonal rearrangements of their T-cell receptor genes. These patients have a higher frequency of skin lesions, more variable lymphocyte shape, and shorter median survival (13 months) with minimal responses to chemotherapy and B-cell receptor inhibitors.[14]
The differential diagnosis must exclude the following:
- Monoclonal B-cell lymphocytosis (MBL). MBL, the precursor to CLL, is defined as a clonal B-cell population circulating in peripheral blood with fewer than 5 × 109 /L B cells and no signs of lymphadenopathy or splenomegaly.[15] Most cases have the immunophenotype of CLL. The incidence of MBL in the general population is 5% to 12% and increases with age.[16] In families with two or more cases of CLL, MBL has a prevalence of 13% to 18%. Low-count MBL (≤0.5 × 109 /L B cells) rarely progresses to overt CLL, but higher levels can progress to symptomatic CLL at a rate of less than 2% per year, even for familial cases.[15,17] In two selected series of more than 900 patients monitored prospectively for a median of 5 to 7 years, overt CLL requiring chemotherapy occurred in 7% of patients.[8,18] A screening study using the Mayo Clinic Biobank identified 1,712 patients with MBL from 10,139 screened samples.[19] Low-count MBL was found in 95% of these patients. With a median follow-up of 10.0 years, only 0.58% of patients progressed to a lymphoid malignancy.[19]
- Hairy cell leukemia. For more information, see Hairy Cell Leukemia Treatment.
- Waldenström macroglobulinemia. Waldenström macroglobulinemia has a natural history and therapeutic options similar to CLL, with the exception of hyperviscosity syndrome associated with macroglobulinemia as a result of elevated immunoglobulin M. For more information, see Indolent B-Cell Non-Hodgkin Lymphoma Treatment.
- Large granular lymphocyte (LGL) leukemia. LGL leukemia is characterized by lymphocytosis with a natural killer (NK) cell immunophenotype (CD2, CD16, and CD56) or a T-cell immunophenotype (CD2, CD3, and CD8).[20,21,22] These patients often have neutropenia and a history of rheumatoid arthritis. The natural history is indolent, often marked by anemia and splenomegaly. This condition appears to fit into the clinical spectrum of Felty syndrome.[23] A characteristic genetic finding in almost 50% of the patients with T-cell LGL involves pathogenic variants in the STAT3 gene.[24] Symptomatic patients with cytopenias typically manifest CD8-positive T cells with alpha/beta surface receptors plus a STAT3 variant, CD8-positive T cells with T gamma/delta surface receptors, or a mutated NK/T-cell phenotype.[25,26] Conversely, asymptomatic patients have wild-type STAT3 CD8-positive T cells, CD4-positive T cells, and wild-type NK cells.[25] Therapy includes low doses of oral cyclophosphamide or methotrexate, cyclosporine, and treatment of the bacterial infections acquired during severe neutropenia.[20,22,27,28]
For information on prolymphocytic leukemia, which was previously covered in this summary, see the Treatment of T-Cell Prolymphocytic Leukemia section in Peripheral T-Cell Non-Hodgkin Lymphoma Treatment.
Prognostic Factors
Prognostic markers help stratify patients in clinical trials, assess the need for therapy, and select the type of therapy.[2,29,30] Prognostic factors that may help predict clinical outcome include cytogenetic subgroup, immunoglobulin mutational status, and CD38 immunophenotype.[2,31,32,33,34,35,36,37,38,39]
Prognostic markers include the following:
- IGH pathogenic variant.[32,33,34,39,40] The finding of significant numbers of variants in this region is associated with a median survival in excess of 20 to 25 years. The absence of variants is associated with a median survival of 8 to 10 years.
- FISH test results. FISH chromosomal abnormalities were associated with prognosis in retrospective and prospective studies and clonal evolution has been seen over time.[31,41,42,43] The following chromosomal abnormalities have been reported:
- del(13q) is a favorable prognostic marker (median overall survival [OS], 17 years in one prospective study).[43]
- Trisomy 12 and del(11) have a less favorable prognosis (median OS, 9–11 years in one prospective study).[43]
- del(17p) is associated with TP53 pathogenic variants, poor response rates, and short duration of response to the standard therapeutic options.[39] del(17p) is associated with the most unfavorable prognosis (median OS, 7 years in one prospective trial).[43,44,45]
- The combination of abnormal cytogenetics, such as del(11q) or del(17p) deletion (suggesting a worse prognosis), with zeta-chain-associated protein 70 kDa negativity (suggesting a better prognosis) in the same patients resulted in a poor prognosis.[38]
These findings emphasize the need for prospective studies of combinations of these prognostic markers.[46]
Other prognostic factors include the following:
- Anemia and thrombocytopenia. These are important adverse prognostic variables, but only if due to extensive marrow involvement by CLL. Autoimmune hemolytic anemia and immune thrombocytopenic purpura do not confer a worse prognosis.
- Age. CLL occurs primarily in middle-aged and older adults, with worse prognosis in successive decades of life.[42]
- Stage.[47,48] For more information, see the sections on the Rai Staging System and the Binet Classification.
- Positron emission tomography (PET)-CT scan results. This test should only be used in the context of recurrent fever, soaking night sweats, weight loss (>10% baseline weight in 6 months), or rapidly growing lymph nodes, because these findings might herald histological transformation to a diffuse large B-cell lymphoma (DLBCL) (so-called Richter transformation). Of 432 patients retrospectively reviewed, 209 patients had a maximum standardized uptake value (SUVmax) of 5 or higher.[49] Eighty percent of these patients had histologically aggressive CLL or Richter syndrome, and both of these entities had equally worse prognoses. When the SUVmax was 10 or higher, the 5-year OS rate was only 30%.[49]
- Lymphocyte doubling time. Doubling of the white blood cell count in less than 1 year implies a worse prognosis.[50]
- Beta-2-microglobulin. Higher levels imply a worse prognosis.[51]
- Richter transformation. In 2% to 10% of patients, CLL will transform into a more aggressive lymphoma, termed Richter transformation.[52] This is usually a DLBCL of the more aggressive activated B-cell subtype. The prognosis is similar to de novo presentations of DLBCL when the CLL has never required therapy or when there is no clonal connection between the CLL and DLBCL (identified if they harbor different clonal light chains or if sequencing can be accomplished at the VDJ [variable, diversity, joining] recombination sites in the immunoglobulin heavy chain variable region).[52] However, the prognosis is poor (median survival, 6–14 months) for most patients with Richter transformation to DLBCL when there has been prior therapy for CLL with chemoimmunotherapy,[53], Bruton tyrosine kinase (BTK) inhibitors, and/or venetoclax.[54] There is no standard therapeutic approach for patients with poor prognoses. Therapies under clinical evaluation include chimeric antigen receptor (CAR) T-cell therapy, T-cell engaging bispecific antibodies, and non-covalent BTK inhibitors.[52,55] Allogeneic stem cell transplant consolidation is often recommended if induction therapy achieves a response.[52] In rare cases of Richter transformation from CLL to Hodgkin lymphoma, the prognosis appears the same for age-matched patients with de novo disease.[56,57]
- Clearance of measurable residual disease (MRD). The improvements in response rates from more intensive regimens have maximized the clearance of MRD. In one prospective trial of 493 patients, clearance of MRD was an independent predictor of OS by multivariate analysis.[58] The surrogate end point of clearance of residual disease, while prognostic,[58,59] did not show improved survival in a randomized prospective trial. The necessary study would include patients who fail to completely clear the marrow with induction therapy and randomly assign them to further alternative treatment versus the same treatment later at relapse, looking at OS as the primary end point.[29,60]
- CD38 immunophenotype.[33,61] CD38 positivity (>30%) correlates with a worse prognosis, but there is a 30% false-positive rate and a 50% false-negative rate using IGH mutational status as the gold standard for prognosis.
- Other malignancies. Patients with CLL are also at increased risk of developing other malignancies, even before therapy.[62] A population-based analysis of almost 2 million cancer patients in the National Cancer Institute's Surveillance, Epidemiology, and End Results (SEER) Program database was performed. The findings suggested that cancer-specific survival for patients with preexisting CLL who subsequently developed colorectal and breast cancer was significantly lower (hazard ratio [HR], 1.46; P < .001 for colorectal cancer and HR, 1.41; P = .005 for breast cancer) than cancer-specific survival for patients with colorectal and breast cancer who did not have antecedent CLL, after adjusting for age, sex, race, and disease stage, and excluding CLL-related deaths.[63]
An international prognostic index (IPI) for CLL (CLL-IPI) identified four prognostic subgroups based on IGH mutational status, clinical stage, age (≤65 years vs. >65 years), and TP53 status (no abnormalities vs. del(17p), TP53 variant, or both).[64] A scoring system to predict time to first treatment for early-stage CLL identified three adverse risk factors: unmutated IGH, absolute lymphocyte count higher than 15 × 109 /L, and palpable lymph nodes.[65] Any new prognostic model, and even the commonly used CLL-IPI, may be outdated because of the use of highly effective frontline therapies, including BCL2 inhibitors and BTK inhibitors.[66] Revalidation of these prognostic models will be required.
Follow-Up After Treatment
CT scans have a very limited role in monitoring patients after completion of treatment. CT scan or ultrasonography results determined the decision to treat for relapse in only 2 of 176 patients in three prospective trials for the German CLL Study Group.[67]
References:
- American Cancer Society: Cancer Facts and Figures 2024. American Cancer Society, 2024. Available online. Last accessed December 30, 2024.
- Burger JA: Treatment of Chronic Lymphocytic Leukemia. N Engl J Med 383 (5): 460-473, 2020.
- Anaissie EJ, Kontoyiannis DP, O'Brien S, et al.: Infections in patients with chronic lymphocytic leukemia treated with fludarabine. Ann Intern Med 129 (7): 559-66, 1998.
- Mauro FR, Foa R, Cerretti R, et al.: Autoimmune hemolytic anemia in chronic lymphocytic leukemia: clinical, therapeutic, and prognostic features. Blood 95 (9): 2786-92, 2000.
- Hallek M, Cheson BD, Catovsky D, et al.: iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 131 (25): 2745-2760, 2018.
- Hallek M, Cheson BD, Catovsky D, et al.: Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 111 (12): 5446-56, 2008.
- Shanafelt TD, Kay NE, Jenkins G, et al.: B-cell count and survival: differentiating chronic lymphocytic leukemia from monoclonal B-cell lymphocytosis based on clinical outcome. Blood 113 (18): 4188-96, 2009.
- Rawstron AC, Bennett FL, O'Connor SJ, et al.: Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 359 (6): 575-83, 2008.
- Dighiero G: Monoclonal B-cell lymphocytosis--a frequent premalignant condition. N Engl J Med 359 (6): 638-40, 2008.
- Fazi C, Scarfò L, Pecciarini L, et al.: General population low-count CLL-like MBL persists over time without clinical progression, although carrying the same cytogenetic abnormalities of CLL. Blood 118 (25): 6618-25, 2011.
- DiGiuseppe JA, Borowitz MJ: Clinical utility of flow cytometry in the chronic lymphoid leukemias. Semin Oncol 25 (1): 6-10, 1998.
- Rozman C, Montserrat E: Chronic lymphocytic leukemia. N Engl J Med 333 (16): 1052-7, 1995.
- Landgren O, Albitar M, Ma W, et al.: B-cell clones as early markers for chronic lymphocytic leukemia. N Engl J Med 360 (7): 659-67, 2009.
- Hoyer JD, Ross CW, Li CY, et al.: True T-cell chronic lymphocytic leukemia: a morphologic and immunophenotypic study of 25 cases. Blood 86 (3): 1163-9, 1995.
- Strati P, Shanafelt TD: Monoclonal B-cell lymphocytosis and early-stage chronic lymphocytic leukemia: diagnosis, natural history, and risk stratification. Blood 126 (4): 454-62, 2015.
- Shim YK, Rachel JM, Ghia P, et al.: Monoclonal B-cell lymphocytosis in healthy blood donors: an unexpectedly common finding. Blood 123 (9): 1319-26, 2014.
- Slager SL, Lanasa MC, Marti GE, et al.: Natural history of monoclonal B-cell lymphocytosis among relatives in CLL families. Blood 137 (15): 2046-2056, 2021.
- Shanafelt TD, Kay NE, Rabe KG, et al.: Brief report: natural history of individuals with clinically recognized monoclonal B-cell lymphocytosis compared with patients with Rai 0 chronic lymphocytic leukemia. J Clin Oncol 27 (24): 3959-63, 2009.
- Slager SL, Parikh SA, Achenbach SJ, et al.: Progression and survival of MBL: a screening study of 10 139 individuals. Blood 140 (15): 1702-1709, 2022.
- Sokol L, Loughran TP: Large granular lymphocyte leukemia. Oncologist 11 (3): 263-73, 2006.
- Semenzato G, Zambello R, Starkebaum G, et al.: The lymphoproliferative disease of granular lymphocytes: updated criteria for diagnosis. Blood 89 (1): 256-60, 1997.
- Lamy T, Loughran TP: How I treat LGL leukemia. Blood 117 (10): 2764-74, 2011.
- Bowman SJ, Sivakumaran M, Snowden N, et al.: The large granular lymphocyte syndrome with rheumatoid arthritis. Immunogenetic evidence for a broader definition of Felty's syndrome. Arthritis Rheum 37 (9): 1326-30, 1994.
- Koskela HL, Eldfors S, Ellonen P, et al.: Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 366 (20): 1905-13, 2012.
- Cheon H, Xing JC, Moosic KB, et al.: Genomic landscape of TCRαβ and TCRγδ T-large granular lymphocyte leukemia. Blood 139 (20): 3058-3072, 2022.
- Barilà G, Grassi A, Cheon H, et al.: Tγδ LGLL identifies a subset with more symptomatic disease: analysis of an international cohort of 137 patients. Blood 141 (9): 1036-1046, 2023.
- Loughran TP, Kidd PG, Starkebaum G: Treatment of large granular lymphocyte leukemia with oral low-dose methotrexate. Blood 84 (7): 2164-70, 1994.
- Dhodapkar MV, Li CY, Lust JA, et al.: Clinical spectrum of clonal proliferations of T-large granular lymphocytes: a T-cell clonopathy of undetermined significance? Blood 84 (5): 1620-7, 1994.
- Developments in the treatment of lymphoproliferative disorders: rising to the new challenges of CLL therapy. A report of a symposium presented during the 48th American Society of Hematology Annual Meeting and Exposition, December 8, 2006, Orlando, Florida. Clin Adv Hematol Oncol 5 (3 Suppl 5): 1-14; quiz 15-6, 2007.
- Pflug N, Bahlo J, Shanafelt TD, et al.: Development of a comprehensive prognostic index for patients with chronic lymphocytic leukemia. Blood 124 (1): 49-62, 2014.
- Döhner H, Stilgenbauer S, Benner A, et al.: Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 343 (26): 1910-6, 2000.
- Hamblin TJ, Davis Z, Gardiner A, et al.: Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 94 (6): 1848-54, 1999.
- Damle RN, Wasil T, Fais F, et al.: Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 94 (6): 1840-7, 1999.
- Rosenwald A, Alizadeh AA, Widhopf G, et al.: Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med 194 (11): 1639-47, 2001.
- Klein U, Tu Y, Stolovitzky GA, et al.: Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med 194 (11): 1625-38, 2001.
- Orchard JA, Ibbotson RE, Davis Z, et al.: ZAP-70 expression and prognosis in chronic lymphocytic leukaemia. Lancet 363 (9403): 105-11, 2004.
- Rassenti LZ, Huynh L, Toy TL, et al.: ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med 351 (9): 893-901, 2004.
- Kröber A, Bloehdorn J, Hafner S, et al.: Additional genetic high-risk features such as 11q deletion, 17p deletion, and V3-21 usage characterize discordance of ZAP-70 and VH mutation status in chronic lymphocytic leukemia. J Clin Oncol 24 (6): 969-75, 2006.
- Byrd JC, Gribben JG, Peterson BL, et al.: Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol 24 (3): 437-43, 2006.
- Kharfan-Dabaja MA, Chavez JC, Khorfan KA, et al.: Clinical and therapeutic implications of the mutational status of IgVH in patients with chronic lymphocytic leukemia. Cancer 113 (5): 897-906, 2008.
- Kröber A, Seiler T, Benner A, et al.: V(H) mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood 100 (4): 1410-6, 2002.
- Catovsky D, Fooks J, Richards S: Prognostic factors in chronic lymphocytic leukaemia: the importance of age, sex and response to treatment in survival. A report from the MRC CLL 1 trial. MRC Working Party on Leukaemia in Adults. Br J Haematol 72 (2): 141-9, 1989.
- Shanafelt TD, Witzig TE, Fink SR, et al.: Prospective evaluation of clonal evolution during long-term follow-up of patients with untreated early-stage chronic lymphocytic leukemia. J Clin Oncol 24 (28): 4634-41, 2006.
- Grever MR, Lucas DM, Dewald GW, et al.: Comprehensive assessment of genetic and molecular features predicting outcome in patients with chronic lymphocytic leukemia: results from the US Intergroup Phase III Trial E2997. J Clin Oncol 25 (7): 799-804, 2007.
- Catovsky D, Richards S, Matutes E, et al.: Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 370 (9583): 230-9, 2007.
- Binet JL, Caligaris-Cappio F, Catovsky D, et al.: Perspectives on the use of new diagnostic tools in the treatment of chronic lymphocytic leukemia. Blood 107 (3): 859-61, 2006.
- Rai KR, Sawitsky A, Cronkite EP, et al.: Clinical staging of chronic lymphocytic leukemia. Blood 46 (2): 219-34, 1975.
- Binet JL, Auquier A, Dighiero G, et al.: A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 48 (1): 198-206, 1981.
- Falchi L, Keating MJ, Marom EM, et al.: Correlation between FDG/PET, histology, characteristics, and survival in 332 patients with chronic lymphoid leukemia. Blood 123 (18): 2783-90, 2014.
- Montserrat E, Sanchez-Bisono J, Viñolas N, et al.: Lymphocyte doubling time in chronic lymphocytic leukaemia: analysis of its prognostic significance. Br J Haematol 62 (3): 567-75, 1986.
- Di Giovanni S, Valentini G, Carducci P, et al.: Beta-2-microglobulin is a reliable tumor marker in chronic lymphocytic leukemia. Acta Haematol 81 (4): 181-5, 1989.
- Parikh SA, Kay NE, Shanafelt TD: How we treat Richter syndrome. Blood 123 (11): 1647-57, 2014.
- Rossi D, Spina V, Deambrogi C, et al.: The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood 117 (12): 3391-401, 2011.
- Kittai AS, Huang Y, Miller S, et al.: Outcomes of patients with Richter transformation without prior chemoimmunotherapy for CLL/SLL: an international multicenter retrospective study. [Abstract] Blood 142 (Suppl 1): A-497, 2023.
- Kittai AS, Bond D, Huang Y, et al.: Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy for Richter Transformation: An International, Multicenter, Retrospective Study. J Clin Oncol 42 (17): 2071-2079, 2024.
- Al-Sawaf O, Robrecht S, Bahlo J, et al.: Richter transformation in chronic lymphocytic leukemia (CLL)-a pooled analysis of German CLL Study Group (GCLLSG) front line treatment trials. Leukemia 35 (1): 169-176, 2021.
- Stephens DM, Boucher K, Kander E, et al.: Hodgkin lymphoma arising in patients with chronic lymphocytic leukemia: outcomes from a large multi-center collaboration. Haematologica 106 (11): 2845-2852, 2021.
- Böttcher S, Ritgen M, Fischer K, et al.: Minimal residual disease quantification is an independent predictor of progression-free and overall survival in chronic lymphocytic leukemia: a multivariate analysis from the randomized GCLLSG CLL8 trial. J Clin Oncol 30 (9): 980-8, 2012.
- Strati P, Keating MJ, O'Brien SM, et al.: Eradication of bone marrow minimal residual disease may prompt early treatment discontinuation in CLL. Blood 123 (24): 3727-32, 2014.
- Montserrat E, Moreno C, Esteve J, et al.: How I treat refractory CLL. Blood 107 (4): 1276-83, 2006.
- Ghia P, Guida G, Stella S, et al.: The pattern of CD38 expression defines a distinct subset of chronic lymphocytic leukemia (CLL) patients at risk of disease progression. Blood 101 (4): 1262-9, 2003.
- Tsimberidou AM, Wen S, McLaughlin P, et al.: Other malignancies in chronic lymphocytic leukemia/small lymphocytic lymphoma. J Clin Oncol 27 (6): 904-10, 2009.
- Solomon BM, Rabe KG, Slager SL, et al.: Overall and cancer-specific survival of patients with breast, colon, kidney, and lung cancers with and without chronic lymphocytic leukemia: a SEER population-based study. J Clin Oncol 31 (7): 930-7, 2013.
- International CLL-IPI working group: An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data. Lancet Oncol 17 (6): 779-90, 2016.
- Condoluci A, Terzi di Bergamo L, Langerbeins P, et al.: International prognostic score for asymptomatic early-stage chronic lymphocytic leukemia. Blood 135 (21): 1859-1869, 2020.
- Kreuzberger N, Damen JA, Trivella M, et al.: Prognostic models for newly-diagnosed chronic lymphocytic leukaemia in adults: a systematic review and meta-analysis. Cochrane Database Syst Rev 7: CD012022, 2020.
- Eichhorst BF, Fischer K, Fink AM, et al.: Limited clinical relevance of imaging techniques in the follow-up of patients with advanced chronic lymphocytic leukemia: results of a meta-analysis. Blood 117 (6): 1817-21, 2011.