




General Information About Childhood Soft Tissue Sarcoma
Dramatic improvements in survival have been achieved for children and adolescents with cancer. Between 1975 and 2020, childhood cancer mortality decreased by more than 50%.[1,2,3] Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. For information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
Pediatric soft tissue sarcomas are a heterogenous group of malignant tumors that originate from primitive mesenchymal tissue and account for 6% of all childhood tumors (rhabdomyosarcomas, 3%; other soft tissue sarcomas, 3%).[2] For more information, see the Histopathological Classification of Childhood Soft Tissue Sarcoma section.
Rhabdomyosarcoma, a tumor of striated muscle, is the most common soft tissue sarcoma in children. It accounts for 50% of the soft tissue sarcomas in children aged 0 to 14 years.[2] For more information, see Childhood Rhabdomyosarcoma Treatment.
In pediatrics, the remaining soft tissue sarcomas are commonly referred to as nonrhabdomyosarcomatous soft tissue sarcomas (NRSTS) and account for approximately 3.5% of all childhood tumors.[2,4] This summary discusses the treatment of NRSTS.
NRSTS are often classified according to the normal tissue types from which they are derived. These types include various connective tissues, peripheral nervous system tissue, smooth muscle tissue, and vascular tissue. The classification also includes undifferentiated tumors that are not clearly related to specific tissue types. For more information about vascular tumors in children, see Childhood Vascular Tumors Treatment.
Incidence of Soft Tissue Sarcoma by Age and Histology
The distribution of soft tissue sarcomas by histology and age, on the basis of the Surveillance, Epidemiology, and End Results (SEER) Program information from 2000 to 2015, is depicted in Table 1. The distribution of histological subtypes by age is also shown in Figure 2.
| | Age <5 y | Age 5–9 y | Age 10–14 y | Age 15–19 y | Age <20 y | All Ages (Including Adults) | ||
|---|---|---|---|---|---|---|---|---|
| pPNET = peripheral primitive neuroectodermal tumors; SEER = Surveillance, Epidemiology, and End Results. | ||||||||
| a Source: SEER database.[5] | ||||||||
| All soft tissue and other extraosseous sarcomas | 1,124 | 773 | 1,201 | 1,558 | 4,656 | 80,269 | ||
| Rhabdomyosarcomas | 668 | 417 | 382 | 327 | 1,794 | 3,284 | ||
| Fibrosarcomas, peripheral nerve, and other fibrous neoplasms | 137 | 64 | 112 | 181 | 494 | 6,645 | ||
| Fibroblastic and myofibroblastic tumors | 114 | 33 | 41 | 77 | 265 | 4,228 | ||
| Nerve sheath tumors | 23 | 31 | 70 | 102 | 226 | 2,303 | ||
| Other fibromatous neoplasms | 0 | 0 | 1 | 2 | 3 | 114 | ||
| Kaposi sarcoma | 2 | 1 | 2 | 10 | 15 | 7,722 | ||
| Other specified soft tissue sarcomas | 237 | 238 | 559 | 865 | 1,899 | 49,004 | ||
| Ewing tumor and Askin tumor of soft tissue | 37 | 36 | 72 | 113 | 258 | 596 | ||
| pPNET of soft tissue | 24 | 23 | 42 | 56 | 145 | 402 | ||
| Extrarenal rhabdoid tumor | 75 | 8 | 9 | 4 | 96 | 205 | ||
| Liposarcomas | 4 | 6 | 37 | 79 | 126 | 10,749 | ||
| Fibrohistiocytic tumors | 43 | 73 | 142 | 223 | 481 | 13,531 | ||
| Leiomyosarcomas | 11 | 14 | 19 | 41 | 85 | 14,107 | ||
| Synovial sarcomas | 12 | 39 | 141 | 210 | 402 | 2,608 | ||
| Blood vessel tumors | 12 | 9 | 11 | 32 | 64 | 4,238 | ||
| Osseous and chondromatous neoplasms of soft tissue | 1 | 6 | 16 | 14 | 37 | 1,018 | ||
| Alveolar soft parts sarcoma | 4 | 5 | 22 | 33 | 64 | 211 | ||
| Miscellaneous soft tissue sarcomas | 14 | 19 | 48 | 60 | 141 | 1,339 | ||
| Unspecified soft tissue sarcomas | 80 | 53 | 146 | 175 | 454 | 13,614 | ||
Soft tissue sarcomas include both rhabdomyosarcomas and NRSTS. NRSTS are more common in adolescents and adults.[6] Most of the information regarding treatment and natural history of the disease in younger patients has been based on studies in adult patients. The distributions of soft tissue sarcomas by age according to stage (Figure 1), histological subtype (Figure 2), and tumor site (Figure 3) are shown below.[7]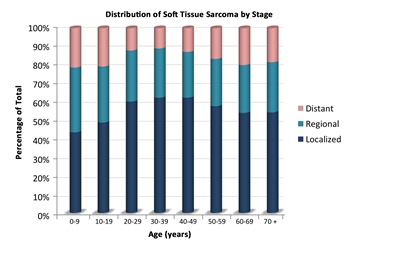
Figure 1. The distribution of soft tissue sarcomas by age according to stage.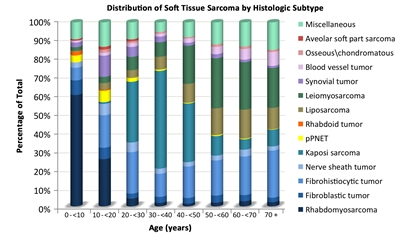
Figure 2. The distribution of soft tissue sarcomas by age according to histological subtype.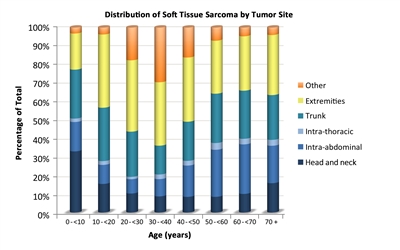
Figure 3. The distribution of soft tissue sarcomas by age according to tumor site.
Risk Factors
Some genetic factors and external exposures have been associated with the development of NRSTS, including the following:
- Genetic factors:
- Li-Fraumeni syndrome: Patients with Li-Fraumeni syndrome (usually resulting from heritable cancer-associated changes of the TP53 tumor suppressor gene) have an increased risk of developing soft tissue tumors (mostly NRSTS), bone sarcomas, breast cancer, brain tumors, and acute leukemia.[8,9]
- Familial adenomatous polyposis: Patients with familial adenomatous polyposis are at increased risk of developing desmoid-type fibromatosis.[10]
- RB1 gene: Germline variants of the RB1 gene have been associated with an increased risk of developing soft tissue sarcoma, particularly leiomyosarcoma, and the risk appears higher among those younger than 1 year who were treated with alkylating agents.[11,12]
- SMARCB1 gene: Germline variants or deletions of the SMARCB1 gene are associated with an increased risk of developing extrarenal rhabdoid tumors.[13] For more information about SMARCB1 and rhabdoid tumor predisposition syndrome type 1, see Rhabdoid Tumor Predisposition Syndrome Type 1.
- Neurofibromatosis type 1: Approximately 4% of patients with neurofibromatosis type 1 develop malignant peripheral nerve sheath tumors, which usually develop after a long latency. Some patients develop multiple lesions.[14,15,16]
- Werner syndrome: Werner syndrome is characterized by spontaneous chromosomal instability, resulting in increased susceptibility to cancer and premature aging. An excess of soft tissue sarcomas has been reported in patients with Werner syndrome.[17]
- Tuberous sclerosis complex: Tuberous sclerosis complex is associated with the development of various tumors showing perivascular epithelioid cell differentiation (PEComas), including lymphangioleiomyomatosis and hepatic and renal angiomyolipomas.[18,19,20]
- Adenosine deaminase–deficient severe combined immunodeficiency: Patients with adenosine deaminase–deficient severe combined immunodeficiency are at increased risk of developing multicentric dermatofibrosarcoma protuberans, which usually presents at an average age of 8.9 years.[21]
- External exposures:
Clinical Presentation
NRSTS can develop in any part of the body, but they arise most commonly in the trunk and extremities.[28,29,30] Although rare, these tumors can arise in brain tissue and are treated according to the histological type.[31]
NRSTS can present initially as an asymptomatic solid mass, or they may be symptomatic because of local invasion or impact on adjacent anatomical structures. Systemic symptoms (e.g., fever, weight loss, and night sweats) are rare. Hypoglycemia and hypophosphatemic rickets have been reported in cases of hemangiopericytoma, which was identified as a solitary fibrous tumor and is now included within myofibroma in the revised World Health Organization (WHO) classification. Hyperglycemia has been noted in patients with fibrosarcoma of the lung.[32]
Diagnostic and Staging Evaluation
When a suspicious lesion is identified, it is crucial to perform a complete workup, followed by adequate biopsy. The lesion is imaged before initiating any intervention using the following procedures:
- Plain films. Plain films can be used to rule out bone involvement and detect calcifications that may be seen in soft tissue tumors such as extraskeletal osteosarcoma or synovial sarcoma.
- Computed tomography (CT). Chest CT is essential to assess the presence of metastases. An abdominal CT can be used to image intra-abdominal tumors, such as liposarcoma. Patients with NRSTS who were treated in 11 centers as part of the European paediatric Soft Tissue Sarcoma Study Group (EpSSG) were retrospectively assessed to evaluate the impact of indeterminate pulmonary nodules identified on chest CT.[33] Of the 206 patients examined, 109 (52.9%) did not have any nodules, 78 (38%) had at least one indeterminate nodule, and 19 (9.2%) had nodules meeting the definition of metastases. The 5-year event-free survival (EFS) rate was 78.5% (95% confidence interval [CI], 69.4%–85.1%) for patients without nodules and 69.6% (95% CI, 57.9%–78.7%) for patients with indeterminate nodules (P = .135). The 5-year overall survival (OS) rate was 87.4% (95% CI, 79.3%–92.5%) for patients without nodules and 79.0% (95% CI, 67.5%–86.8%) for patients with indeterminate nodules (P = .086).
- Magnetic resonance imaging (MRI). MRI may be essential for a surgeon to achieve adequate surgical margins. MRI can be used to image intra-abdominal tumors, such as liposarcoma, and is essential for extremity lesions.
- Positron emission tomography (PET) scan and bone scan. In a retrospective study, 46 PET scans were completed in 25 pediatric patients with soft tissue sarcoma.[34] The positive predictive value of finding metastatic disease was 89%, and the negative predictive value was 67%. A small retrospective study of nine patients with NRSTS suggested that PET-CT was more accurate and cost-effective than either modality alone in identifying distant metastatic disease.[35] The use of this modality in pediatric NRSTS has not been studied prospectively.
The imaging characteristics of some tumors can be highly suggestive of that particular diagnosis. For example, the imaging characteristics of pediatric low-grade fibromyxoid sarcoma and alveolar soft part sarcoma have been described and can aid in the diagnosis of these rare neoplasms.[36]
Biopsy strategies
Although NRSTS are pathologically distinct from rhabdomyosarcoma and Ewing sarcoma, the classification of childhood NRSTS type is often difficult. Core-needle biopsy, incisional biopsy, or excisional biopsy can be used to diagnose NRSTS. If possible, the surgeon who will perform the definitive resection needs to be involved in the biopsy decision. Poorly placed incisional or needle biopsies may adversely affect the ability to achieve negative margins.
Needle biopsy techniques must ensure adequate tissue sampling. Given the diagnostic importance of translocations and other molecular changes, a core-needle biopsy or small incisional biopsy that obtains adequate tumor tissue is crucial to allow for conventional histological and immunocytochemical analysis and other studies such as light and electron microscopy, cytogenetics, fluorescence in situ hybridization, and molecular pathology.[37,38]
The acquisition of multiple cores of tissue may be required. Of 530 suspected soft tissue masses in (largely adult) patients who underwent core-needle biopsies, 426 (80%) were proven to be soft tissue tumors, 225 (52.8%) of which were malignant. Core-needle biopsy was able to differentiate soft tissue sarcomas from benign lesions with a sensitivity of 96.3% and a specificity of 99.4%. Tumor subtype was accurately assigned in 89.5% of benign lesions and in 88% of soft tissue sarcomas. The biopsy complication rate was 0.4%.[39]
Considerations related to a biopsy procedure are as follows:
- Core-needle biopsy for a deep-seated tumor can lead to formation of a hematoma, which affects subsequent resection and/or radiation (because the hematoma should be covered in the irradiated volume).
- Fine-needle biopsy is usually not recommended because it is difficult to determine the accurate histological diagnosis and grade of the tumor in this heterogeneous group of tumors.
- Image guidance using ultrasonography, CT scan, or MRI may be necessary to ensure a representative biopsy.[40] Image guidance is particularly helpful in deep lesions and to avoid cystic changes or necrotic tumors.[41]
- Incisional biopsies must not compromise subsequent wide local excision.
- Excisional biopsy of the lesion is only appropriate for small superficial lesions (<3 cm in size) and are discouraged.[42,43] If an excisional biopsy is contemplated, then MRI of the area is recommended to define the area of involvement as subsequent surgery or radiation therapy may be needed.
- Various institutional series have demonstrated the feasibility and effectiveness of sentinel lymph node biopsy as a staging procedure in pediatric patients with soft tissue sarcomas.[44,45,46,47,48,49] The utility of sentinel node biopsy is currently limited to epithelioid sarcoma, clear cell sarcoma, and rhabdomyosarcoma of the trunk and extremities.[50]
In a prospective study of pediatric patients with sarcoma who underwent sentinel lymph node biopsy, 28 patients were examined. Sentinel lymph node biopsy was positive in 7 of the 28 patients, including 3 patients (43%) who had negative PET-CT scans. PET-CT overestimated and suggested nodal involvement in 14 patients, more than what was confirmed by sentinel lymph node biopsy. The findings from the sentinel lymph node biopsies resulted in altering therapy for all seven patients who were determined to have metastatic disease. As indicated by previous reports, epithelioid sarcoma and clear cell sarcoma were the two NRSTS included in this study.[50]
- In the ARST0332 (NCT00346164) study, patients with epithelioid sarcoma, clear cell sarcoma, or radiographically enlarged nodes underwent regional node sampling. Nodal metastases were identified in 20 patients (3.8%), and all but one of these patients had radiographic evidence of nodal involvement. The most common histologies included epithelioid sarcoma (18%), angiosarcoma (17%), and clear cell sarcoma (14%). Patients with isolated nodal metastases had a similar outcome to those who did not have distant metastases (5-year OS rates, 85% vs. 87%). Sentinel lymph node biopsies were encouraged but not required for this study. A sentinel lymph node biopsy was not done in most patients because they had clinically enlarged nodes. Of note, three patients without clinical evidence of lymph node metastasis at study entry experienced lymph node basin failure. One of these patients had three lymph nodes in two different lymph node basins sampled by sentinel lymph node biopsy that were pathologically negative.[51]
Transverse extremity incisions are avoided to reduce skin loss at re-excision and because they require a greater cross-sectional volume of tissue to be covered in the radiation field. Other extensive surgical procedures are also avoided before definitive diagnosis.
For these reasons, open biopsy or multiple core-needle biopsies are strongly encouraged so that adequate tumor tissue can be obtained to allow crucial studies to be performed and to avoid limiting future treatment options.
Unplanned resection
In children with unplanned resection of NRSTS, primary re-excision is frequently recommended because many patients will have tumor present in the re-excision specimen.[52,53] A single-institution analysis of adolescents and adults compared patients who had unplanned excisions of soft tissue sarcoma to stage-matched controls. In this retrospective analysis, unplanned initial excision of soft tissue sarcoma resulted in increased risk of local recurrence, metastasis, and death. This increased risk was greatest for high-grade tumors.[54][Level of evidence C1] In this case, a second resection is expected.
Chromosomal abnormalities
Many NRSTS are characterized by chromosomal abnormalities. Some of these chromosomal translocations lead to a fusion of two disparate genes. The resulting fusion transcript can be readily detected by using polymerase chain reaction–based techniques, thus facilitating the diagnosis of those neoplasms that have translocations.
Some of the most frequent aberrations seen in NRSTS are listed in Table 2.
| Histology | Chromosomal Aberrations | Genes Involved |
|---|---|---|
| a Adapted from Sandberg,[55]Slater et al.,[56]Mertens et al.,[57]Romeo,[58]and Schaefer et al.[59] | ||
| Alveolar soft part sarcoma | t(x;17)(p11.2;q25) | ASPSCR1::TFE3[60,61,62] |
| Angiomatoid fibrous histiocytoma | t(12;16)(q13;p11), t(2;22)(q33;q12), t(12;22)(q13;q12) | FUS::ATF1,EWSR1::CREB1,[63]EWSR1::ATF1 |
| BCOR-rearranged sarcomas | inv(X)(p11.4;p11.2) | BCOR::CCNB3 |
| CIC-rearranged sarcomas | t(4;19)(q35;q13), t(10;19)(q26;q13) | CIC::DUX4 |
| Clear cell sarcoma | t(12;22)(q13;q12), t(2;22)(q33;q12) | EWSR1::ATF1,EWSR1::CREB1[64] |
| Congenital (infantile) fibrosarcoma/mesoblastic nephroma | t(12;15)(p13;q25) | ETV6::NTRK3 |
| Dermatofibrosarcoma protuberans | t(17;22)(q22;q13) | COL1A1::PDGFB |
| Desmoid fibromatosis | Trisomy 8 or 20, loss of 5q21 | CTNNB1orAPCvariants |
| Desmoplastic small round cell tumors | t(11;22)(p13;q12) | EWSR1::WT1[65,66] |
| Epithelioid hemangioendothelioma | t(1;3)(p36;q25)[67] | WWTR1::CAMTA1 |
| Epithelioid sarcoma | Inactivation ofSMARCB1 | SMARCB1 |
| Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12), t(9;17)(q22;q11), t(9;15)(q22;q21), t(3;9)(q11;q22) | EWSR1::NR4A3,TAF2N::NR4A3,TCF12::NR4A3,TFG::NR4A3 |
| Hemangiopericytoma (myofibroma) | t(12;19)(q13;q13.3) and t(13;22)(q22;q13.3) | LMNA::NTRK1[68] |
| Infantile fibrosarcoma | t(12;15)(p13;q25) | ETV6::NTRK3 |
| Inflammatory myofibroblastic tumor | t(1;2)(q23;q23), t(2;19)(q23;q13), t(2;17)(q23;q23), t(2;2)(p23;q13), t(2;11)(p23;p15)[69] | TPM3::ALK,TPM4::ALK,CLTC::ALK,RANBP2::ALK,CARS1::ALK,RAS |
| Infantile myofibromatosis | Gain-of-function variants | PDGFRB[70] |
| Low-grade fibromyxoid sarcoma | t(7;16)(q33;p11), t(11;16)(p11;p11) | FUS::CREB3L2,FUS::CREB3L1 |
| Malignant peripheral nerve sheath tumor | 17q11.2, loss or rearrangement of 10p, 11q, 17q, 22q | NF1 |
| Mesenchymal chondrosarcoma | Del(8)(q13.3q21.1) | HEY1::NCOA2 |
| Myoepithelioma | t(19;22)(q13;q12), t(1;22)(q23;q12), t(6;22)(p21;q12) | EWSR1::ZNF44,EWSR1::PBX1,EWSR1::POU5F1 |
| Myxoid/round cell liposarcoma | t(12;16)(q13;p11), t(12;22)(q13;q12) | FUS::DDIT3,EWSR1::DDIT3 |
| Primitive myxoid mesenchymal tumor of infancy | Internal tandem duplication | BCOR |
| Rhabdoid tumor | Inactivation ofSMARCB1 | SMARCB1 |
| Sclerosing epithelioid fibrosarcoma | t(11;22)(p11;q12), t(19;22)(p13;q12) | EWSR1::CREB3L1,EWSR1::CREB3L3 |
| Solitary fibrous tumor | inv(12)(q13q13) | NAB2::STAT6 |
| Synovial sarcoma | t(x;18)(p11.2;q11.2) | SS18::SSX |
| Tenosynovial giant cell tumor | t(1;2)(p13;q35) | COL6A3::CSF1 |
Prognosis and Prognostic Factors
The prognosis of NRSTS varies greatly depending on the following factors:[71,72,73]
- Site of the primary tumor.
- Tumor size.
- Tumor grade. For more information, see the Soft Tissue Sarcoma Tumor Pathological Grading System section.
- Tumor histology.
- Depth of tumor invasion.
- Presence of metastases and site of the metastatic tumor.
- Resectability of the tumor.
- Use of radiation therapy.
In a review of a large adult series of NRSTS, patients with superficial extremity sarcomas had a better prognosis than did patients with deep tumors. This may be a reflection of differences in resectability. Thus, in addition to grade and size, the depth of invasion of the tumor should be considered.[74]
Data specific to NRSTS in children and adolescents are difficult to separate. Several adult and pediatric series have shown that patients with large or invasive tumors have a significantly worse prognosis than do those with small, noninvasive tumors. A retrospective review of soft tissue sarcomas (rhabdomyosarcoma and NRSTS) in children and adolescents suggests that the 5-cm cutoff used for adults with soft tissue sarcoma may not be ideal for smaller children, especially infants. The review identified an interaction between tumor diameter and body surface area.[75] This relationship has been questioned in a rhabdomyosarcoma study and requires further study to determine the therapeutic implications of the observation.[76]
Some pediatric NRSTS are associated with a better outcome. For instance, patients with infantile fibrosarcoma who present at age 4 years or younger have an excellent prognosis. This excellent outcome occurs because surgery alone can cure a significant number of these patients and infantile fibrosarcoma is highly chemosensitive. This tumor also responds well to larotrectinib, a specific tropomyosin receptor kinase inhibitor.[22,77]
Prognosis based on the Children's Oncology Group (COG) ARST0332 trial
Soft tissue sarcomas in older children and adolescents often behave similarly to those in adult patients.[22,78] A large, prospective, multinational COG study (ARST0332 [NCT00346164]) enrolled newly diagnosed patients younger than 30 years. Patients were assigned to treatment based on their risk group. Risk groups were defined by the presence of metastasis, tumor resectability and margins, and tumor size and grade. For more information, see Figure 4.[79][Level of evidence B4]
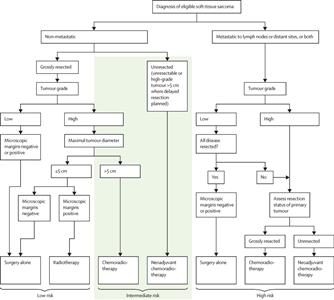
Figure 4. Risk group and treatment assignment for the Children's Oncology Group ARST0332 trial. Reprinted from The Lancet Oncology, Volume 21 (Issue 1), Spunt SL, Million L, Chi YY, et al., A risk-based treatment strategy for non-rhabdomyosarcoma soft-tissue sarcomas in patients younger than 30 years (ARST0332): a Children's Oncology Group prospective study, Pages 145–161, Copyright © 2020, with permission from Elsevier.
Each patient was assigned to one of three risk groups and one of four treatment groups. The risk groups were as follows:[79]
- Low risk: Nonmetastatic R0 (resection was complete with negative microscopic margins) or R1 (microscopically positive margins) low-grade tumor, or ≤5 cm R1 high-grade tumor.
- Intermediate risk: Nonmetastatic R0 or R1 >5 cm high-grade tumor, or unresected tumor of any size or grade.
- High risk: Metastatic tumor.
The treatment groups were as follows:
- Surgery alone (n = 205).
- Radiation therapy (55.8 Gy) (n = 17).
- Chemoradiation therapy (chemotherapy and 55.8 Gy radiation therapy) (n = 111).
- Neoadjuvant chemoradiation therapy (chemotherapy and 45 Gy radiation therapy, then surgery and radiation therapy boost based on margins with continued chemotherapy) (n = 196).
Chemotherapy included six cycles of ifosfamide (3 g/m2 per dose) given intravenously on days 1 through 3 and five cycles of doxorubicin (37.5 mg/m2 per dose) given intravenously on days 1 to 2 every 3 weeks, with the sequence adjusted based on the timing of surgery or radiation therapy.
For the 550 patients enrolled, 529 evaluable patients were included in the analysis. At a median follow-up of 6.5 years (interquartile range [IQR], 4.9–7.9), the survival results are shown in Table 3.
| | 5-Year Event-Free Survival | 5-Year Overall Survival | ||
|---|---|---|---|---|
| Risk Group | Events/Patients | Estimate, % (95% CI) | Events/Patients | Estimate, % (95% CI) |
| CI = confidence interval; R0 = completely excised with negative microscopic margins; R1 = grossly excised but with positive microscopic margins; R2 = less than complete gross excision. | ||||
| Low | 26/222 | 88.9 (84.0–93.8) | 10/222 | 96.2 (93.2–99.2) |
| Intermediate | 84/227 | 65.0 (58.2–71.8) | 55/227 | 79.2 (73.4–85.0) |
| High | 63/80 | 21.2 (11.4–31.1) | 52/80 | 35.5 (23.6–47.4) |
| Surgical Margin | ||||
| R0 | 44/252 | 83.6 (78.3–89.0) | 22/252 | 92.8 (89.1–96.5) |
| R1 | 29/81 | 66.2 (54.8–77.5) | 17/81 | 79.7 (70.0–89.5) |
| R2 | 100/196 | 49.2 (41.4–57.0) | 78/196 | 62.7 (55.2–70.3) |
The COG ARST0332 trial was a risk-based stratification study. Overall, local control after radiation therapy was as follows: R0, 106 of 109 patients (97%); R1, 51 of 60 patients (85%); and R2/unresectable, 2 of 6 patients (33%). Local recurrence predictors included extent of delayed resection (P < .001), imaging response before delayed surgery (P < .001), histological subtype (P < .001), and no radiation therapy (P = .046). The 5-year EFS was significantly lower for patients unable to undergo R0 or R1 resection (P = .0003).[80]
Pediatric patients with unresected localized NRSTS have a poor outcome. Only about one-third of patients treated with multimodality therapy remain disease free.[71,81]; [82,83][Level of evidence C1] In an Italian review of 30 patients with NRSTS at visceral sites, only ten patients survived at 5 years. Unfavorable prognostic factors included inability to achieve complete resection, large tumor size, tumor invasion, histological subtype, and lung-pleura sites.[84][Level of evidence C1]
Prognosis based on the European paediatric Soft Tissue Sarcoma Study Group (EpSSG) NRSTS 2005 study
The EpSSG conducted a prospective trial for patients younger than 21 years with NRSTS. They reported an analysis of 206 patients with synovial sarcoma and 363 with adult-type NRSTS. Patients were treated according to assigned risk groups. For more information, see Figure 5.[85] With a median follow-up of 80 months (interquartile range, 54.3–111.3) for the 467 surviving patients, the 5-year EFS rate was 73.7% (95% CI, 69.7%–77.2%), and the OS rate was 83.8% (95% CI, 80.3%–86.7%). The survival by treatment groups are shown in Table 4.[85]
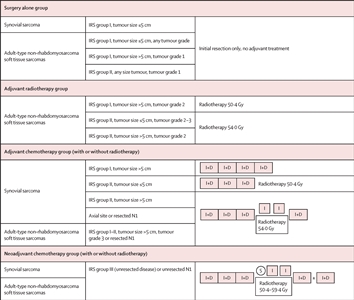
Figure 5. Treatment plan for patients with synovial sarcoma or adult-type non-rhabdomyosarcoma soft tissue sarcomas. Patients were divided into four treatment groups based on surgical stage, tumour size, nodal involvement, tumour grade (according to the Fédération Nationale des Centres de Lutte Contre le Cancer grading system for adult-type non-rhabdomyosarcoma soft tissue sarcomas), and tumour site (for synovial sarcoma). I+D = ifosfamide (3.0 g/m2 per day intravenously for 3 days) plus doxorubicin (37.5 mg/m2 per day intravenously for 2 days). I = ifosfamide (3.0 g/m2 per day intravenously for 2 days). IRS = Intergroup Rhabdomyosarcoma Study. N1 = nodal involvement. S = delayed surgery. Reprinted from The Lancet Child & Adolescent Health, Volume 5, Issue 8, Ferrari A, van Noesel MM, Brennan B, et al., Paediatric non-rhabdomyosarcoma soft tissue sarcomas: the prospective NRSTS 2005 study by the European paediatric Soft Tissue Sarcoma Study Group (EpSSG), Pages 546-558, Copyright 2021, with permission from Elsevier.
| Treatment Group | 5-Year Event-Free Survival Rate (95% CI) | 5-Year Overall Survival Rate (95% CI) | Local Recurrence Rate |
|---|---|---|---|
| CI = confidence interval; EpSSG = European paediatric Soft Tissue Sarcoma Study Group; NRSTS = nonrhabdomyosarcomatous soft tissue sarcomas. | |||
| Surgery alone | 91.4% (87.0%–94.4%) | 98.1% (95.0%–99.3%) | 7.6% (19/250) |
| Adjuvant radiation therapy alone (n = 17) | 75.5% (46.9%–90.1%) | 88.2% (60.6%–96.9%) | 6.7% (1/15) |
| Adjuvant chemotherapy ± radiation therapy (n = 93) | 65.6% (54.8%–74.5%) | 75.8% (65.3%–83.5%) | 10.8% (7/65) |
| Neoadjuvant chemotherapy ± radiation therapy (n = 209) | 56.4% (49.3%–63.0%) | 70.4% (63.3%–76.4%) | 14.2% (16/113) |
Treatment failures specifically for the neoadjuvant therapy treatment groups are shown in Table 5.[85]
| Treatment | Local Failure (No. of Patients) | Local + Metastatic Failure (No. of Patients) | Metastatic Failure (No. of Patients) |
|---|---|---|---|
| EpSSG = European paediatric Soft Tissue Sarcoma Study Group; No. = number; NRSTS = nonrhabdomyosarcomatous soft tissue sarcomas. | |||
| a Adapted from Ferrari et al.[85] | |||
| Radiation therapy alone (n = 21) | 7 | 2 | 4 |
| Delayed surgery followed by radiation therapy (n = 104) | 16 | 6 | 8 |
| Delayed surgery alone (n = 48) | 8 | 3 | 8 |
| No local treatment (n = 16) | 12 | 4 | 0 |
| Preoperative radiation therapy followed by delayed surgery (n = 20) | 4 | 0 | 6 |
The authors concluded that adjuvant therapy (radiation therapy and chemotherapy) could safely be omitted in the group of patients assigned to surgery alone. Their criteria included the following:[85]
- Synovial cell: Intergroup Rhabdomyosarcoma Study (IRS) group I tumor size <5 cm.
- Adult-type NRSTS: IRS group I tumor size <5 cm, any grade.
- Adult-type NRSTS: IRS group I tumor size >5 cm, tumor grade I.
- Adult-type NRSTS: IRS group II any tumor size, tumor grade I.
They also concluded that improving the outcome for patients with high-risk, initially resected, adult-type NRSTS and those with initially unresected disease remains a major clinical challenge.[85]
In a pooled analysis from U.S. and European pediatric centers, outcomes were better for patients whose tumor removal procedure was deemed complete than for patients whose tumor removal was incomplete. Outcomes were better for patients who received radiation therapy than for patients who did not.[82][Level of evidence C1]
Because long-term morbidity must be minimized while disease-free survival is maximized, the ideal therapy for each patient must be carefully and individually determined using these prognostic factors before initiating therapy.[29,86,87,88,89,90]
References:
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed August 23, 2024.
- Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed September 5, 2024.
- Hawkins DS, Black JO, Orbach D, et al.: Nonrhabdomyosarcoma soft-tissue sarcomas. In: Blaney SM, Helman LJ, Adamson PC, eds.: Pizzo and Poplack's Pediatric Oncology. 8th ed. Wolters Kluwer, 2020, pp 721-46.
- Surveillance, Epidemiology, and End Results (SEER) Program: SEER*Stat Database: Incidence - SEER 18 Regs Research Data + Hurricane Katrina Impacted Louisiana Cases, Nov 2017 Sub (1973-2015 varying) - Linked To County Attributes - Total U.S., 1969-2016 Counties [Database]. National Cancer Institute, DCCPS, Surveillance Research Program, released April 2018, based on the November 2017 submission. Available online. Last accessed October 12, 2022.
- Weiss SW, Goldblum JR: General considerations. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. Mosby, 2008, pp 1-14.
- Ferrari A, Sultan I, Huang TT, et al.: Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer 57 (6): 943-9, 2011.
- Chang F, Syrjänen S, Syrjänen K: Implications of the p53 tumor-suppressor gene in clinical oncology. J Clin Oncol 13 (4): 1009-22, 1995.
- Plon SE, Malkin D: Childhood cancer and hereditary. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Lippincott Williams and Wilkins, 2015, pp 13-31.
- Groen EJ, Roos A, Muntinghe FL, et al.: Extra-intestinal manifestations of familial adenomatous polyposis. Ann Surg Oncol 15 (9): 2439-50, 2008.
- Kleinerman RA, Tucker MA, Abramson DH, et al.: Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst 99 (1): 24-31, 2007.
- Wong JR, Morton LM, Tucker MA, et al.: Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. J Clin Oncol 32 (29): 3284-90, 2014.
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011.
- Weiss SW, Goldblum JR: Benign tumors of peripheral nerves. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. Mosby, 2008, pp 825-901.
- deCou JM, Rao BN, Parham DM, et al.: Malignant peripheral nerve sheath tumors: the St. Jude Children's Research Hospital experience. Ann Surg Oncol 2 (6): 524-9, 1995.
- Stark AM, Buhl R, Hugo HH, et al.: Malignant peripheral nerve sheath tumours--report of 8 cases and review of the literature. Acta Neurochir (Wien) 143 (4): 357-63; discussion 363-4, 2001.
- Goto M, Miller RW, Ishikawa Y, et al.: Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 5 (4): 239-46, 1996.
- Fricke BL, Donnelly LF, Casper KA, et al.: Frequency and imaging appearance of hepatic angiomyolipomas in pediatric and adult patients with tuberous sclerosis. AJR Am J Roentgenol 182 (4): 1027-30, 2004.
- Adriaensen ME, Schaefer-Prokop CM, Duyndam DA, et al.: Radiological evidence of lymphangioleiomyomatosis in female and male patients with tuberous sclerosis complex. Clin Radiol 66 (7): 625-8, 2011.
- Hornick JL, Fletcher CD: PEComa: what do we know so far? Histopathology 48 (1): 75-82, 2006.
- Kesserwan C, Sokolic R, Cowen EW, et al.: Multicentric dermatofibrosarcoma protuberans in patients with adenosine deaminase-deficient severe combined immune deficiency. J Allergy Clin Immunol 129 (3): 762-769.e1, 2012.
- Spunt SL, Million L, Coffin C: The nonrhabdomyosarcoma soft tissue sarcoma. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Lippincott Williams and Wilkins, 2015, pp 827-54.
- Weiss SW, Goldblum JR: Malignant fibrous histiocytoma (pleomorphic undifferentiated sarcoma). In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. Mosby, 2008, pp 403-27.
- Tukenova M, Guibout C, Hawkins M, et al.: Radiation therapy and late mortality from second sarcoma, carcinoma, and hematological malignancies after a solid cancer in childhood. Int J Radiat Oncol Biol Phys 80 (2): 339-46, 2011.
- Bartkowiak D, Humble N, Suhr P, et al.: Second cancer after radiotherapy, 1981-2007. Radiother Oncol 105 (1): 122-6, 2012.
- Casey DL, Friedman DN, Moskowitz CS, et al.: Second cancer risk in childhood cancer survivors treated with intensity-modulated radiation therapy (IMRT). Pediatr Blood Cancer 62 (2): 311-316, 2015.
- McClain KL, Leach CT, Jenson HB, et al.: Association of Epstein-Barr virus with leiomyosarcomas in children with AIDS. N Engl J Med 332 (1): 12-8, 1995.
- Dillon P, Maurer H, Jenkins J, et al.: A prospective study of nonrhabdomyosarcoma soft tissue sarcomas in the pediatric age group. J Pediatr Surg 27 (2): 241-4; discussion 244-5, 1992.
- Rao BN: Nonrhabdomyosarcoma in children: prognostic factors influencing survival. Semin Surg Oncol 9 (6): 524-31, 1993 Nov-Dec.
- Zeytoonjian T, Mankin HJ, Gebhardt MC, et al.: Distal lower extremity sarcomas: frequency of occurrence and patient survival rate. Foot Ankle Int 25 (5): 325-30, 2004.
- Benesch M, von Bueren AO, Dantonello T, et al.: Primary intracranial soft tissue sarcoma in children and adolescents: a cooperative analysis of the European CWS and HIT study groups. J Neurooncol 111 (3): 337-45, 2013.
- Weiss SW, Goldblum JR: Miscellaneous tumors of intermediate malignancy. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. Mosby, 2008, pp 1093-1160.
- Giraudo C, Schoot R, Cardoen L, et al.: Indeterminate pulmonary nodules in non-rhabdomyosarcoma soft tissue sarcoma: A study of the European paediatric Soft Tissue Sarcoma Study Group. Cancer 130 (4): 597-608, 2024.
- Mody RJ, Bui C, Hutchinson RJ, et al.: FDG PET imaging of childhood sarcomas. Pediatr Blood Cancer 54 (2): 222-7, 2010.
- Tateishi U, Hosono A, Makimoto A, et al.: Accuracy of 18F fluorodeoxyglucose positron emission tomography/computed tomography in staging of pediatric sarcomas. J Pediatr Hematol Oncol 29 (9): 608-12, 2007.
- Sargar K, Kao SC, Spunt SL, et al.: MRI and CT of Low-Grade Fibromyxoid Sarcoma in Children: A Report From Children's Oncology Group Study ARST0332. AJR Am J Roentgenol 205 (2): 414-20, 2015.
- Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. Mosby, 2008.
- Recommendations for the reporting of soft tissue sarcomas. Association of Directors of Anatomic and Surgical Pathology. Mod Pathol 11 (12): 1257-61, 1998.
- Strauss DC, Qureshi YA, Hayes AJ, et al.: The role of core needle biopsy in the diagnosis of suspected soft tissue tumours. J Surg Oncol 102 (5): 523-9, 2010.
- Chowdhury T, Barnacle A, Haque S, et al.: Ultrasound-guided core needle biopsy for the diagnosis of rhabdomyosarcoma in childhood. Pediatr Blood Cancer 53 (3): 356-60, 2009.
- Tuttle R, Kane JM: Biopsy techniques for soft tissue and bowel sarcomas. J Surg Oncol 111 (5): 504-12, 2015.
- Coffin CM, Dehner LP, O'Shea PA: Pediatric Soft Tissue Tumors: A Clinical, Pathological, and Therapeutic Approach. Williams and Wilkins, 1997.
- Smith LM, Watterson J, Scott SM: Medical and surgical management of pediatric soft tissue tumors. In: Coffin CM, Dehner LP, O'Shea PA: Pediatric Soft Tissue Tumors: A Clinical, Pathological, and Therapeutic Approach. Williams and Wilkins, 1997, pp 360-71.
- Neville HL, Andrassy RJ, Lally KP, et al.: Lymphatic mapping with sentinel node biopsy in pediatric patients. J Pediatr Surg 35 (6): 961-4, 2000.
- Neville HL, Raney RB, Andrassy RJ, et al.: Multidisciplinary management of pediatric soft-tissue sarcoma. Oncology (Huntingt) 14 (10): 1471-81; discussion 1482-6, 1489-90, 2000.
- Kayton ML, Delgado R, Busam K, et al.: Experience with 31 sentinel lymph node biopsies for sarcomas and carcinomas in pediatric patients. Cancer 112 (9): 2052-9, 2008.
- Dall'Igna P, De Corti F, Alaggio R, et al.: Sentinel node biopsy in pediatric patients: the experience in a single institution. Eur J Pediatr Surg 24 (6): 482-7, 2014.
- Parida L, Morrisson GT, Shammas A, et al.: Role of lymphoscintigraphy and sentinel lymph node biopsy in the management of pediatric melanoma and sarcoma. Pediatr Surg Int 28 (6): 571-8, 2012.
- Alcorn KM, Deans KJ, Congeni A, et al.: Sentinel lymph node biopsy in pediatric soft tissue sarcoma patients: utility and concordance with imaging. J Pediatr Surg 48 (9): 1903-6, 2013.
- Wagner LM, Kremer N, Gelfand MJ, et al.: Detection of lymph node metastases in pediatric and adolescent/young adult sarcoma: Sentinel lymph node biopsy versus fludeoxyglucose positron emission tomography imaging-A prospective trial. Cancer 123 (1): 155-160, 2017.
- Alvarez E, He J, Spunt SL, et al.: Lymph node metastases in paediatric and young adult patients with non-rhabdomyosarcoma soft tissue sarcoma (NRSTS): Findings from Children's Oncology Group (COG) study ARST0332. Eur J Cancer 180: 89-98, 2023.
- Chui CH, Spunt SL, Liu T, et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg 37 (10): 1424-9, 2002.
- Cecchetto G, Guglielmi M, Inserra A, et al.: Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int 17 (7): 532-4, 2001.
- Qureshi YA, Huddy JR, Miller JD, et al.: Unplanned excision of soft tissue sarcoma results in increased rates of local recurrence despite full further oncological treatment. Ann Surg Oncol 19 (3): 871-7, 2012.
- Sandberg AA: Translocations in malignant tumors. Am J Pathol 159 (6): 1979-80, 2001.
- Slater O, Shipley J: Clinical relevance of molecular genetics to paediatric sarcomas. J Clin Pathol 60 (11): 1187-94, 2007.
- Mertens F, Antonescu CR, Hohenberger P, et al.: Translocation-related sarcomas. Semin Oncol 36 (4): 312-23, 2009.
- Romeo S, Dei Tos AP: Clinical application of molecular pathology in sarcomas. Curr Opin Oncol 23 (4): 379-84, 2011.
- Schaefer IM, Cote GM, Hornick JL: Contemporary Sarcoma Diagnosis, Genetics, and Genomics. J Clin Oncol 36 (2): 101-110, 2018.
- Ladanyi M, Lui MY, Antonescu CR, et al.: The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 20 (1): 48-57, 2001.
- Ladanyi M: The emerging molecular genetics of sarcoma translocations. Diagn Mol Pathol 4 (3): 162-73, 1995.
- Williams A, Bartle G, Sumathi VP, et al.: Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: useful diagnostic tools in cases with unusual histological features. Virchows Arch 458 (3): 291-300, 2011.
- Antonescu CR, Dal Cin P, Nafa K, et al.: EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 46 (12): 1051-60, 2007.
- Hisaoka M, Ishida T, Kuo TT, et al.: Clear cell sarcoma of soft tissue: a clinicopathologic, immunohistochemical, and molecular analysis of 33 cases. Am J Surg Pathol 32 (3): 452-60, 2008.
- Barnoud R, Sabourin JC, Pasquier D, et al.: Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: a comparative study with other small round cell tumors. Am J Surg Pathol 24 (6): 830-6, 2000.
- Wang LL, Perlman EJ, Vujanic GM, et al.: Desmoplastic small round cell tumor of the kidney in childhood. Am J Surg Pathol 31 (4): 576-84, 2007.
- Errani C, Zhang L, Sung YS, et al.: A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 50 (8): 644-53, 2011.
- Haller F, Knopf J, Ackermann A, et al.: Paediatric and adult soft tissue sarcomas with NTRK1 gene fusions: a subset of spindle cell sarcomas unified by a prominent myopericytic/haemangiopericytic pattern. J Pathol 238 (5): 700-10, 2016.
- Jain S, Xu R, Prieto VG, et al.: Molecular classification of soft tissue sarcomas and its clinical applications. Int J Clin Exp Pathol 3 (4): 416-28, 2010.
- Agaimy A, Bieg M, Michal M, et al.: Recurrent Somatic PDGFRB Mutations in Sporadic Infantile/Solitary Adult Myofibromas But Not in Angioleiomyomas and Myopericytomas. Am J Surg Pathol 41 (2): 195-203, 2017.
- Spunt SL, Hill DA, Motosue AM, et al.: Clinical features and outcome of initially unresected nonmetastatic pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Clin Oncol 20 (15): 3225-35, 2002.
- Spunt SL, Poquette CA, Hurt YS, et al.: Prognostic factors for children and adolescents with surgically resected nonrhabdomyosarcoma soft tissue sarcoma: an analysis of 121 patients treated at St Jude Children's Research Hospital. J Clin Oncol 17 (12): 3697-705, 1999.
- Ferrari A, Casanova M, Collini P, et al.: Adult-type soft tissue sarcomas in pediatric-age patients: experience at the Istituto Nazionale Tumori in Milan. J Clin Oncol 23 (18): 4021-30, 2005.
- Brooks AD, Heslin MJ, Leung DH, et al.: Superficial extremity soft tissue sarcoma: an analysis of prognostic factors. Ann Surg Oncol 5 (1): 41-7, 1998 Jan-Feb.
- Ferrari A, Miceli R, Meazza C, et al.: Soft tissue sarcomas of childhood and adolescence: the prognostic role of tumor size in relation to patient body size. J Clin Oncol 27 (3): 371-6, 2009.
- Rodeberg DA, Stoner JA, Garcia-Henriquez N, et al.: Tumor volume and patient weight as predictors of outcome in children with intermediate risk rhabdomyosarcoma: a report from the Children's Oncology Group. Cancer 117 (11): 2541-50, 2011.
- Hong DS, DuBois SG, Kummar S, et al.: Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol 21 (4): 531-540, 2020.
- Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 4th ed. Mosby, 2001.
- Spunt SL, Million L, Chi YY, et al.: A risk-based treatment strategy for non-rhabdomyosarcoma soft-tissue sarcomas in patients younger than 30 years (ARST0332): a Children's Oncology Group prospective study. Lancet Oncol 21 (1): 145-161, 2020.
- Million L, Hayes-Jordan A, Chi YY, et al.: Local Control For High-Grade Nonrhabdomyosarcoma Soft Tissue Sarcoma Assigned to Radiation Therapy on ARST0332: A Report From the Childrens Oncology Group. Int J Radiat Oncol Biol Phys 110 (3): 821-830, 2021.
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002.
- Ferrari A, Miceli R, Rey A, et al.: Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Cancer 47 (5): 724-31, 2011.
- Smith KB, Indelicato DJ, Knapik JA, et al.: Definitive radiotherapy for unresectable pediatric and young adult nonrhabdomyosarcoma soft tissue sarcoma. Pediatr Blood Cancer 57 (2): 247-51, 2011.
- Ferrari A, Magni C, Bergamaschi L, et al.: Pediatric nonrhabdomyosarcoma soft tissue sarcomas arising at visceral sites. Pediatr Blood Cancer 64 (9): , 2017.
- Ferrari A, van Noesel MM, Brennan B, et al.: Paediatric non-rhabdomyosarcoma soft tissue sarcomas: the prospective NRSTS 2005 study by the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG). Lancet Child Adolesc Health 5 (8): 546-558, 2021.
- Dillon PW, Whalen TV, Azizkhan RG, et al.: Neonatal soft tissue sarcomas: the influence of pathology on treatment and survival. Children's Cancer Group Surgical Committee. J Pediatr Surg 30 (7): 1038-41, 1995.
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994.
- Marcus KC, Grier HE, Shamberger RC, et al.: Childhood soft tissue sarcoma: a 20-year experience. J Pediatr 131 (4): 603-7, 1997.
- Pratt CB, Pappo AS, Gieser P, et al.: Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 17 (4): 1219, 1999.
- Pratt CB, Maurer HM, Gieser P, et al.: Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol 30 (4): 201-9, 1998.