




General Information About Childhood Extracranial Germ Cell Tumors (GCTs)
GCTs arise from primordial germ cells, which migrate during embryogenesis from the yolk sac through the mesentery to the gonads (see Figure 1).[1,2] Childhood extracranial GCTs can generally be divided into gonadal and extragonadal. These tumors can also be broadly classified as teratomas, malignant GCTs, or mixed GCTs.
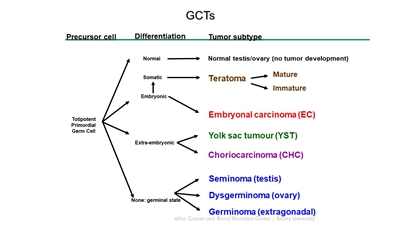
Figure 1. Extracranial germ cell development from primordial germ cells. Credit: Thomas Olson, M.D.
Incidence
Childhood GCTs are rare in children younger than 15 years, accounting for approximately 3% of cancers in this age group.[3,4,5,6] In the fetal/neonatal age group, most extracranial GCTs are benign teratomas occurring at midline locations, including the head and neck, sacrococcyx, and retroperitoneum.[7,8] Despite the small percentage of malignant teratomas that occur in this age group, perinatal tumors have a high morbidity rate caused by hydrops fetalis and premature delivery.[8,9,10]
The incidence of malignant extracranial GCTs increases with the onset of puberty. These tumors represent approximately 15% of cancers in male adolescents aged 15 to 19 years and 4% of cancers in female adolescents aged 15 to 19 years.[3]
Figure 2 shows the age-incidence profile by sex for malignant extracranial/extragonadal GCTs (left panel) and malignant gonadal GCTs (right panel) between 2014 and 2018 for 23 U.S. Cancer registries that represent 66% of all U.S. children, adolescents, and young adults (blue triangles, females; green triangles, males).[3] For males, there is a peak in incidence in children younger than 2 years for both extragonadal and gonadal sites, which is followed by low rates between the ages of 2 and 12 years, and then higher rates throughout adolescence. For females, the peak in young children is present only for extragonadal tumors, with rates increasing after the age of puberty for both extragonadal and gonadal sites. However, the incidence of each tumor is lower for females during adolescence than for males during adolescence.
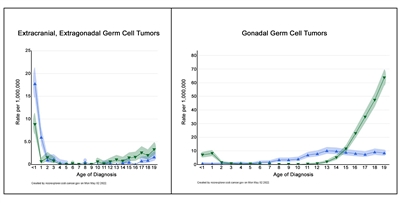
Figure 2. Age-incidence profiles for extracranial, extragonadal germ cell tumors (left graph) and for gonadal germ cell tumors (right graph). The blue triangles represent females, and the green triangles represent males. (See text for details.)
The incidence of extracranial GCTs according to age group, sex, and gonadal versus extragonadal primary site is shown in Table 1.[3]
| Tumor Site | Sex | Age <1 y | Ages 1–4 y | Ages 5–9 y | Ages 10–14 y | Ages 15–19 y |
|---|---|---|---|---|---|---|
| a Rates are per 1 million children from 2014 to 2018 for NCCR Registries, 23 U.S. Cancer registries that represent 66% of all U.S. children, adolescents, and young adults. | ||||||
| b Data from National Cancer Institute; National Childhood Cancer Registry: NCCR*Explorer.[3] | ||||||
| Extragonadal | Female | 17.7 | 2.1 | 0.1 | 0.1 | 0.7 |
| Male | 8.8 | 0.7 | 0 | 0.6 | 2.2 | |
| Gonadal | Female | 0.6 | 0.7 | 2.1 | 7.6 | 8.3 |
| Male | 7 | 2.5 | 0.1 | 1.5 | 36.1 | |
Risk Factors
Cryptorchidism, the presence of an abdominal undescended testis, has been associated with a 10.8-fold increased risk of developing a GCT.[11] Gonadal dysgenesis, as well as the presence of Y-chromosome material in an abdominal gonad, also increases the risk of developing a gonadal GCT, especially gonadoblastoma. Gonadoblastoma is a rare gonadal tumor consisting of a mixture of germ cells and sex-cord stromal derivatives resembling immature granulosa and Sertoli cells.[12,13]
There are few data about the potential genetic or environmental risk factors associated with childhood extragonadal extracranial GCTs. Patients with the following syndromes are at an increased risk of extragonadal extracranial GCTs:
- Klinefelter syndrome: Increased risk of mediastinal GCTs.[14,15,16,17]
Most mediastinal GCTs in adolescents and young adults occur in males, and 22% to 50% have cytogenetic changes consistent with Klinefelter syndrome.[15,18] The age of tumor presentation is younger in patients with Klinefelter syndrome, and testing all younger males for Klinefelter syndrome should be considered.[15,18]
Patients with GCTs were identified from the Children's Oncology Group (COG) Childhood Cancer Research Network. Twenty-nine patients in the study had mediastinal primary tumors, and nine patients (31%) had Klinefelter syndrome. In the Centers for Disease Control and Prevention's large 2013 WONDER database, 3% of patients with GCTs had Klinefelter syndrome (70% were mediastinal). In comparison, 0.2% of males in the general population have Klinefelter syndrome.[17]
- Swyer syndrome: Increased risk of gonadoblastomas and seminomas.[19,20]
- Turner syndrome: Increased risk of gonadoblastomas and dysgerminomas.[21,22]
Histological Classification of Childhood Extracranial GCTs
Childhood extracranial GCTs comprise a variety of histological diagnoses and can be broadly classified as the following:
- Teratomas.
- Malignant GCTs.
The histological properties of extracranial GCTs are heterogeneous and vary by primary tumor site and the sex and age of the patient.[23,24] Histologically identical GCTs that arise in younger children have different biological characteristics from those that arise in adolescents and young adults.[25]
Mature teratoma
Mature teratomas can occur at gonadal or at extragonadal locations. They are the most common histological subtype of childhood GCT.[10,26,27,28] Mature teratomas usually contain well-differentiated tissues from the ectodermal, mesodermal, and endodermal germ cell layers. Any tissue type may be found within this tumor.
Mature teratomas are benign, although some mature teratomas may secrete enzymes or hormones, including insulin, growth hormone, androgens, and prolactin.[29,30]
Immature teratoma
Immature teratomas contain tissues from the ectodermal, mesodermal, and endodermal germ cell layers. Immature tissues, primarily neuroepithelial, are also present. Immature teratomas are graded from 0 to 3 on the basis of the amount of immature neural tissue found in the tumor specimen.[31,32] Tumors of higher grade are more likely to have foci of yolk sac tumor.[33] Immature teratomas can exhibit malignant behavior and metastasize.
Immature teratomas occur primarily in young children at extragonadal sites and in the ovaries of girls near the age of puberty. However, there is no correlation between tumor grade and patient age.[33,34] Some immature teratomas may secrete enzymes or hormones such as vasopressin.[35]
Malignant GCTs
Most childhood extragonadal GCTs arise in midline sites (i.e., head and neck, sacrococcygeal, mediastinal, and retroperitoneal). The midline location may represent aberrant embryonic migration of the primordial germ cells.
GCTs contain malignant tissues of germ cell origin and, rarely, tissues of somatic origin. Isolated malignant elements may constitute a small fraction of a predominantly mature or immature teratoma.[34,36]
Malignant germ cell elements of children, adolescents, and young adults can be grouped broadly by location (see Table 2).
| Malignant Germ Cell Elements | Location | |
|---|---|---|
| E = extragonadal; O = ovarian; T = testicular. | ||
| a Modified from Perlman et al.[37] | ||
| Seminomatous | ||
| Seminoma | T | |
| Dysgerminoma | O | |
| Germinoma | E | |
| Nonseminomatous | ||
| Yolk sac tumor (endodermal sinus tumor) | E, O, T | |
| Choriocarcinoma | E, O, T | |
| Embryonal carcinoma | E, T | |
| Gonadoblastoma | O | |
| Mixed Germ Cell Tumors | ||
| Mixed germ cell tumors | E, O, T | |
GCT Biology
Childhood extracranial GCTs develop at many sites, including testicles, ovaries, mediastinum, retroperitoneum, sacrum, coccyx, and head and neck (see Figure 3).[7] The clinical features at presentation are specific for each site.
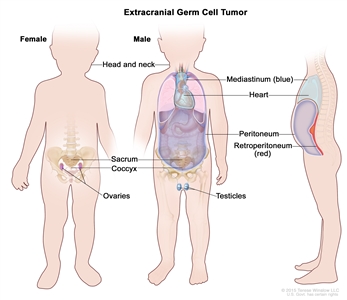
Figure 3. Extracranial germ cell tumors form in parts of the body other than the brain. This includes the testicles, ovaries, sacrococcyx (usually originating from the coccyx and including the sacrum), mediastinum, and retroperitoneum.
The following biologically distinct subtypes of GCTs are found in children and adolescents:
Biological distinctions between GCTs in children and GCTs in adults may not be absolute, and biological factors have not been shown to predict risk.[38,39,40]
Testicular GCTs
- Children (aged <11 years): During early childhood, both testicular teratomas and malignant testicular GCTs are identified. The malignant tumors are commonly composed of pure yolk sac tumor (also known as endodermal sinus tumor) and are generally diploid or tetraploid. Up to approximately 44% of testicular GCTs contain the isochromosome of the short arm of chromosome 12 (i12p) that characterizes testicular cancer in young adults.[38,41,42,43,44,45] Deletions of chromosomes 1p, 4q, and 6q and gains of chromosomes 1q, 3, and 20q are reported as recurring chromosomal abnormalities for this group of tumors.[43,44,45,46]
- Adolescents and young adults (aged ≥11 years): Testicular GCTs in the adolescent and young adult population almost always possess an i12p chromosomal abnormality [47,48,49,50] and are aneuploid.[41,50]
Ovarian GCTs
Ovarian GCTs occur primarily in adolescent and young adult females. While most ovarian GCTs are benign mature teratomas (dermoid cysts), a heterogeneous group of malignant GCTs, including immature teratomas, dysgerminomas, yolk sac tumors, and mixed GCTs, do occur in females. The malignant ovarian GCT commonly shows increased copies of the short arm of chromosome 12.[51]
Extragonadal extracranial GCTs
Extragonadal extracranial GCTs occur outside of the brain and gonads.
- Children (aged <11 years): These tumors typically present at birth or during early childhood. Most of these tumors are benign teratomas occurring in the sacrococcygeal region, and thus are not included in Surveillance, Epidemiology, and End Results (SEER) Program data.[52,53] Malignant yolk sac tumor histology occurs in a minority of these tumors; however, they may have cytogenetic abnormalities similar to those observed for tumors occurring in the testes of young males.[42,43,44,46] Mediastinal GCTs in children younger than 8 years share the same genetic gains and losses as do sacrococcygeal and testicular tumors in young children.[18,54,55]
- Older children, adolescents, and young adults (aged ≥11 years): The mediastinum is the most common primary site for extragonadal GCTs in older children and adolescents.[27]
For information about the treatment of intracranial GCTs, see Childhood Central Nervous System Germ Cell Tumors Treatment.
Diagnostic and Staging Evaluation
Diagnostic evaluation of GCTs includes measurement of serum tumor markers and imaging studies. In suspected cases, tumor markers can suggest the diagnosis before surgery and/or biopsy. This information can be used by the multidisciplinary team to make appropriate treatment choices.
Tumor markers
Tumor markers are measured with each cycle of chemotherapy for all pediatric patients with malignant GCTs. After initial chemotherapy, tumor markers may show a transient elevation.[56]
Common tumor markers include the following:
- Alpha-fetoprotein (AFP).
The fetal liver produces AFP, and during the first year of life, infants have elevated serum AFP levels, which are not associated with the presence of a GCT. Normal ranges have been described.[57,58] The serum half-life of AFP is 5 to 7 days.
Yolk sac tumors produce AFP. Most children with malignant GCTs will have a component of yolk sac tumor and have elevations of AFP levels,[59,60] which are serially monitored during treatment to help assess response to therapy.[34,36,59] Benign teratomas and immature teratomas may produce small elevations of AFP and beta-human chorionic gonadotropin (beta-hCG).
A COG study measured AFP levels in children who received chemotherapy for GCTs. AFP decline was defined as automatically satisfactory if AFP normalized after two cycles of chemotherapy and was calculated satisfactory if the AFP half-life decline was less than or equal to 7 days after the start of chemotherapy. Other decline in AFP was defined as unsatisfactory.[61][Level of evidence C1]
- The cumulative incidence of relapse was 11% for patients with a satisfactory decline in AFP (n = 117) and 38% for patients with an unsatisfactory decline in AFP (n = 14).
- Beta-hCG.
Beta-hCG is produced by all choriocarcinomas and by some germinomas (seminomas and dysgerminomas) and embryonal carcinomas, resulting in elevated serum levels of these substances. The serum half-life of beta-hCG is 1 to 2 days.
- MicroRNAs.
In a prospective multicentric study, the serum level of microRNA-371a-3p was shown to be a sensitive and specific biomarker for adult testicular GCTs.[62] The study included 616 patients with GCTs of varying histologies and 258 controls without malignant GCTs. Elevation of microRNA-371a-3p levels was noted in all malignant histologies, including seminomas. Normal controls and patients with benign teratomas did not have the biomarker elevation. MicroRNA-371a-3p levels were related to tumor volume, and the levels decreased in response to chemotherapy. More studies about microRNA-371a-3p are needed to assess its use in patients with pediatric GCTs.
Imaging tests
Imaging tests may include the following:
- Computed tomography (CT) scan of the chest.
- CT or magnetic resonance imaging (MRI) of the primary site.
- Radionuclide bone scan, if clinically indicated.
- MRI of the brain, if clinically indicated.
Prognostic Factors
Prognostic factors for extracranial GCTs depend on many patient and tumor characteristics and include the following (obtained from historical national GCT trials):[59,63,64,65]
- Age (e.g., young children vs. adolescents).
- Stage of disease.
- Primary site of disease.
- Histology (e.g., seminomatous vs. nonseminomatous).
- Tumor marker decline (AFP and beta-hCG) in response to therapy.
- Presence of gonadal dysgenesis.
To better identify prognostic factors, data from five U.S. trials and two U.K. trials for malignant extracranial GCTs in children and adolescents were merged by the Malignant Germ Cell Tumor International Collaborative. The goal was to ascertain the important prognostic factors in 519 young patients who received chemotherapy, incorporating age at diagnosis, stage, and site of primary tumor, along with pretreatment AFP level and histology.[66][Level of evidence C2] In this age-focused investigation of these factors in young children and adolescents, outcomes included the following (see Figure 4):[66]
- Patients aged 11 years and older with stage III or stage IV extragonadal disease or stage IV ovarian disease had a less than 70% likelihood of long-term disease-free survival, ranging from 40% (extragonadal stage IV) to 67% (ovarian stage IV).
- Boys (aged 11 years and older) with International Germ Cell Consensus Classification [67] intermediate-risk or poor-risk features also had inferior outcomes.
- Presence of a yolk sac tumor predicted better outcome, but it did not achieve statistical significance at the .05 level.
- Preoperative AFP levels were not prognostic. Postoperative AFP levels were prognostic in adult men.[67]
A subsequent study used a database of 11 GCT trials and identified 593 patients with metastatic testicular, mediastinal, or retroperitoneal GCTs. The distribution of patients by age groups included 90 children (aged 0 to <11 years), 109 adolescents (aged 11 to <18 years), and 394 young adults (aged 18 to ≤30 years).[67]; [68][Level of evidence C1]
- The 5-year event-free survival (EFS) rate was lower for adolescents (72%; 95% confidence interval [CI], 62%–79%) than it was for children (90%; 95% CI, 81%–95%; P = .003) or young adults (88%; 95% CI, 84%–91%; P = .0002).
- After adjusting for the International Germ Cell Consensus Classification risk group,[67] only the difference in EFS between adolescents and children remained significant (hazard ratio, 0.30; P = .001).
Although few pediatric data exist, adult studies have shown that an unsatisfactory decline of elevated tumor markers after the first cycle of chemotherapy is a poor prognostic finding.[69,70]
The presence of gonadal dysgenesis in patients with ovarian nondysgerminomas is associated with worse outcomes. In a report from the COG AGCT0132 study, seven patients with gonadal dysgenesis and ovarian nondysgerminomas had an estimated 3-year EFS rate of 67%, compared with 89% for 100 patients with nondysgerminoma ovarian tumors who did not have gonadal dysgenesis.[13] These dysgenetic gonads contain Y-chromosome material, and intra-abdominal gonads with Y-chromosome material are at increased risk of tumor development.[12,71] In contrast to nondysgerminomas, gonadal dysgenesis was identified in 7 of 48 patients with ovarian dysgerminomas in a report from the French Society of Pediatric Oncology. With a medium follow-up of 14 years, all patients survived.[72]
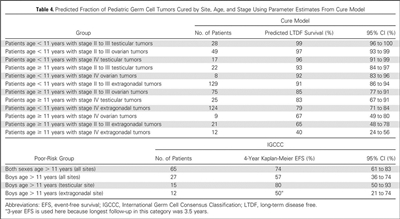
Figure 4. Predicted fraction of pediatric germ cell tumors cured by site, age, and stage using parameter estimates from cure model. Reprinted with permission. © 2015 American Society of Clinical Oncology. All rights reserved. Frazier AL, Hale JP, Rodriguez-Galindo C, et al: Revised risk classification for pediatric extracranial germ cell tumors based on 25 years of clinical trial data from the United Kingdom and United States. J Clin Oncol, Vol. 33 (Issue 2), 2015: 195-201.
For more information about prognosis and prognostic factors for childhood extragonadal extracranial GCTs, see the sections on Treatment of Mature and Immature Teratomas in Children, Treatment of Malignant Gonadal GCTs in Children, and Treatment of Malignant Extragonadal Extracranial GCTs in Children.
Follow-up After Treatment
The following tests and procedures may be performed at the physician's discretion for monitoring children with extracranial GCTs:
- AFP and beta-hCG. Monitor AFP and beta-hCG levels monthly for 6 months (period of highest risk) and then every 3 months, for a total of 2 years (3 years for sacrococcygeal teratoma).
A COG trial of patients with low-risk and intermediate-risk GCTs reported the following results:[73][Level of evidence C2]
- Forty-eight patients with elevated tumor markers at diagnosis relapsed during the surveillance phase.
- At the time of relapse (after central review), 47 of 48 (98%) relapses were detected by tumor marker elevation.
- Imaging tests.
- MRI/CT may be performed at the completion of therapy.
- Guided imaging of the primary site may be performed every 3 months for the first year and every six months for the second year. Seminomas and dysgerminomas may recur later, so the imaging schedule may need to be extended.
- Chest x-ray annually.
- When tumor markers are normal at diagnosis, ultrasonography or CT/MRI may be performed every 3 months for 2 years and then annually for 5 years for germinomas.
Dramatic improvements in survival have been achieved for children and adolescents with cancer.[74] Between 1975 and 2020, childhood cancer mortality decreased by more than 50%.[3,74,75] During the period from 2002 to 2010, cancer mortality continued to decrease by 2.4% per year for children and adolescents with gonadal tumors, as compared with the period from 1975 to 1998 (plateauing from 1998 to 2001).[74] Childhood and adolescent cancer survivors require close monitoring because late effects of cancer therapy may persist or develop months or years after treatment. For information about the incidence, type, and monitoring of late effects of childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
References:
- Dehner LP: Gonadal and extragonadal germ cell neoplasia of childhood. Hum Pathol 14 (6): 493-511, 1983.
- McIntyre A, Gilbert D, Goddard N, et al.: Genes, chromosomes and the development of testicular germ cell tumors of adolescents and adults. Genes Chromosomes Cancer 47 (7): 547-57, 2008.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed August 23, 2024.
- Poynter JN, Amatruda JF, Ross JA: Trends in incidence and survival of pediatric and adolescent patients with germ cell tumors in the United States, 1975 to 2006. Cancer 116 (20): 4882-91, 2010.
- Kaatsch P, Häfner C, Calaminus G, et al.: Pediatric germ cell tumors from 1987 to 2011: incidence rates, time trends, and survival. Pediatrics 135 (1): e136-43, 2015.
- Ward E, DeSantis C, Robbins A, et al.: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 64 (2): 83-103, 2014 Mar-Apr.
- Dharmarajan H, Rouillard-Bazinet N, Chandy BM: Mature and immature pediatric head and neck teratomas: A 15-year review at a large tertiary center. Int J Pediatr Otorhinolaryngol 105: 43-47, 2018.
- Isaacs H: Perinatal (fetal and neonatal) germ cell tumors. J Pediatr Surg 39 (7): 1003-13, 2004.
- Heerema-McKenney A, Harrison MR, Bratton B, et al.: Congenital teratoma: a clinicopathologic study of 22 fetal and neonatal tumors. Am J Surg Pathol 29 (1): 29-38, 2005.
- Alexander VR, Manjaly JG, Pepper CM, et al.: Head and neck teratomas in children--A series of 23 cases at Great Ormond Street Hospital. Int J Pediatr Otorhinolaryngol 79 (12): 2008-14, 2015.
- Johnson KJ, Ross JA, Poynter JN, et al.: Paediatric germ cell tumours and congenital abnormalities: a Children's Oncology Group study. Br J Cancer 101 (3): 518-21, 2009.
- Huang H, Wang C, Tian Q: Gonadal tumour risk in 292 phenotypic female patients with disorders of sex development containing Y chromosome or Y-derived sequence. Clin Endocrinol (Oxf) 86 (4): 621-627, 2017.
- Dicken BJ, Billmire DF, Krailo M, et al.: Gonadal dysgenesis is associated with worse outcomes in patients with ovarian nondysgerminomatous tumors: A report of the Children's Oncology Group AGCT 0132 study. Pediatr Blood Cancer 65 (4): , 2018.
- Dexeus FH, Logothetis CJ, Chong C, et al.: Genetic abnormalities in men with germ cell tumors. J Urol 140 (1): 80-4, 1988.
- Nichols CR, Heerema NA, Palmer C, et al.: Klinefelter's syndrome associated with mediastinal germ cell neoplasms. J Clin Oncol 5 (8): 1290-4, 1987.
- Lachman MF, Kim K, Koo BC: Mediastinal teratoma associated with Klinefelter's syndrome. Arch Pathol Lab Med 110 (11): 1067-71, 1986.
- Williams LA, Pankratz N, Lane J, et al.: Klinefelter syndrome in males with germ cell tumors: A report from the Children's Oncology Group. Cancer 124 (19): 3900-3908, 2018.
- Schneider DT, Schuster AE, Fritsch MK, et al.: Genetic analysis of mediastinal nonseminomatous germ cell tumors in children and adolescents. Genes Chromosomes Cancer 34 (1): 115-25, 2002.
- Coutin AS, Hamy A, Fondevilla M, et al.: [Pure 46XY gonadal dysgenesis] J Gynecol Obstet Biol Reprod (Paris) 25 (8): 792-6, 1996.
- Amice V, Amice J, Bercovici JP, et al.: Gonadal tumor and H-Y antigen in 46,XY pure gonadal dysgenesis. Cancer 57 (7): 1313-7, 1986.
- Tanaka Y, Sasaki Y, Tachibana K, et al.: Gonadal mixed germ cell tumor combined with a large hemangiomatous lesion in a patient with Turner's syndrome and 45,X/46,X, +mar karyotype. Arch Pathol Lab Med 118 (11): 1135-8, 1994.
- Kota SK, Gayatri K, Pani JP, et al.: Dysgerminoma in a female with turner syndrome and Y chromosome material: A case-based review of literature. Indian J Endocrinol Metab 16 (3): 436-40, 2012.
- Hawkins EP: Germ cell tumors. Am J Clin Pathol 109 (4 Suppl 1): S82-8, 1998.
- Schneider DT, Calaminus G, Koch S, et al.: Epidemiologic analysis of 1,442 children and adolescents registered in the German germ cell tumor protocols. Pediatr Blood Cancer 42 (2): 169-75, 2004.
- Horton Z, Schlatter M, Schultz S: Pediatric germ cell tumors. Surg Oncol 16 (3): 205-13, 2007.
- Göbel U, Calaminus G, Engert J, et al.: Teratomas in infancy and childhood. Med Pediatr Oncol 31 (1): 8-15, 1998.
- Rescorla FJ: Pediatric germ cell tumors. Semin Surg Oncol 16 (2): 144-58, 1999.
- Harms D, Zahn S, Göbel U, et al.: Pathology and molecular biology of teratomas in childhood and adolescence. Klin Padiatr 218 (6): 296-302, 2006 Nov-Dec.
- Tomlinson MW, Alaverdian AA, Alaverdian V: Testosterone-producing benign cystic teratoma with virilism. A case report. J Reprod Med 41 (12): 924-6, 1996.
- Kallis P, Treasure T, Holmes SJ, et al.: Exocrine pancreatic function in mediastinal teratomata: an aid to preoperative diagnosis? Ann Thorac Surg 54 (4): 741-3, 1992.
- Norris HJ, Zirkin HJ, Benson WL: Immature (malignant) teratoma of the ovary: a clinical and pathologic study of 58 cases. Cancer 37 (5): 2359-72, 1976.
- O'Connor DM, Norris HJ: The influence of grade on the outcome of stage I ovarian immature (malignant) teratomas and the reproducibility of grading. Int J Gynecol Pathol 13 (4): 283-9, 1994.
- Heifetz SA, Cushing B, Giller R, et al.: Immature teratomas in children: pathologic considerations: a report from the combined Pediatric Oncology Group/Children's Cancer Group. Am J Surg Pathol 22 (9): 1115-24, 1998.
- Marina NM, Cushing B, Giller R, et al.: Complete surgical excision is effective treatment for children with immature teratomas with or without malignant elements: A Pediatric Oncology Group/Children's Cancer Group Intergroup Study. J Clin Oncol 17 (7): 2137-43, 1999.
- Lam SK, Cheung LP: Inappropriate ADH secretion due to immature ovarian teratoma. Aust N Z J Obstet Gynaecol 36 (1): 104-5, 1996.
- Göbel U, Calaminus G, Schneider DT, et al.: The malignant potential of teratomas in infancy and childhood: the MAKEI experiences in non-testicular teratoma and implications for a new protocol. Klin Padiatr 218 (6): 309-14, 2006 Nov-Dec.
- Perlman EJ, Hawkins EP: Pediatric germ cell tumors: protocol update for pathologists. Pediatr Dev Pathol 1 (4): 328-35, 1998 Jul-Aug.
- Palmer RD, Foster NA, Vowler SL, et al.: Malignant germ cell tumours of childhood: new associations of genomic imbalance. Br J Cancer 96 (4): 667-76, 2007.
- Palmer RD, Barbosa-Morais NL, Gooding EL, et al.: Pediatric malignant germ cell tumors show characteristic transcriptome profiles. Cancer Res 68 (11): 4239-47, 2008.
- Poynter JN, Hooten AJ, Frazier AL, et al.: Associations between variants in KITLG, SPRY4, BAK1, and DMRT1 and pediatric germ cell tumors. Genes Chromosomes Cancer 51 (3): 266-71, 2012.
- Oosterhuis JW, Castedo SM, de Jong B, et al.: Ploidy of primary germ cell tumors of the testis. Pathogenetic and clinical relevance. Lab Invest 60 (1): 14-21, 1989.
- Silver SA, Wiley JM, Perlman EJ: DNA ploidy analysis of pediatric germ cell tumors. Mod Pathol 7 (9): 951-6, 1994.
- Perlman EJ, Cushing B, Hawkins E, et al.: Cytogenetic analysis of childhood endodermal sinus tumors: a Pediatric Oncology Group study. Pediatr Pathol 14 (4): 695-708, 1994 Jul-Aug.
- Schneider DT, Schuster AE, Fritsch MK, et al.: Genetic analysis of childhood germ cell tumors with comparative genomic hybridization. Klin Padiatr 213 (4): 204-11, 2001 Jul-Aug.
- Bussey KJ, Lawce HJ, Olson SB, et al.: Chromosome abnormalities of eighty-one pediatric germ cell tumors: sex-, age-, site-, and histopathology-related differences--a Children's Cancer Group study. Genes Chromosomes Cancer 25 (2): 134-46, 1999.
- Perlman EJ, Valentine MB, Griffin CA, et al.: Deletion of 1p36 in childhood endodermal sinus tumors by two-color fluorescence in situ hybridization: a pediatric oncology group study. Genes Chromosomes Cancer 16 (1): 15-20, 1996.
- Rodriguez E, Houldsworth J, Reuter VE, et al.: Molecular cytogenetic analysis of i(12p)-negative human male germ cell tumors. Genes Chromosomes Cancer 8 (4): 230-6, 1993.
- Bosl GJ, Ilson DH, Rodriguez E, et al.: Clinical relevance of the i(12p) marker chromosome in germ cell tumors. J Natl Cancer Inst 86 (5): 349-55, 1994.
- Mostert MC, Verkerk AJ, van de Pol M, et al.: Identification of the critical region of 12p over-representation in testicular germ cell tumors of adolescents and adults. Oncogene 16 (20): 2617-27, 1998.
- van Echten J, Oosterhuis JW, Looijenga LH, et al.: No recurrent structural abnormalities apart from i(12p) in primary germ cell tumors of the adult testis. Genes Chromosomes Cancer 14 (2): 133-44, 1995.
- Riopel MA, Spellerberg A, Griffin CA, et al.: Genetic analysis of ovarian germ cell tumors by comparative genomic hybridization. Cancer Res 58 (14): 3105-10, 1998.
- Malogolowkin MH, Mahour GH, Krailo M, et al.: Germ cell tumors in infancy and childhood: a 45-year experience. Pediatr Pathol 10 (1-2): 231-41, 1990.
- Marsden HB, Birch JM, Swindell R: Germ cell tumours of childhood: a review of 137 cases. J Clin Pathol 34 (8): 879-83, 1981.
- Dal Cin P, Drochmans A, Moerman P, et al.: Isochromosome 12p in mediastinal germ cell tumor. Cancer Genet Cytogenet 42 (2): 243-51, 1989.
- Aly MS, Dal Cin P, Jiskoot P, et al.: Competitive in situ hybridization in a mediastinal germ cell tumor. Cancer Genet Cytogenet 73 (1): 53-6, 1994.
- Vogelzang NJ, Lange PH, Goldman A, et al.: Acute changes of alpha-fetoprotein and human chorionic gonadotropin during induction chemotherapy of germ cell tumors. Cancer Res 42 (11): 4855-61, 1982.
- Wu JT, Book L, Sudar K: Serum alpha fetoprotein (AFP) levels in normal infants. Pediatr Res 15 (1): 50-2, 1981.
- Blohm ME, Vesterling-Hörner D, Calaminus G, et al.: Alpha 1-fetoprotein (AFP) reference values in infants up to 2 years of age. Pediatr Hematol Oncol 15 (2): 135-42, 1998 Mar-Apr.
- Mann JR, Raafat F, Robinson K, et al.: The United Kingdom Children's Cancer Study Group's second germ cell tumor study: carboplatin, etoposide, and bleomycin are effective treatment for children with malignant extracranial germ cell tumors, with acceptable toxicity. J Clin Oncol 18 (22): 3809-18, 2000.
- Marina N, Fontanesi J, Kun L, et al.: Treatment of childhood germ cell tumors. Review of the St. Jude experience from 1979 to 1988. Cancer 70 (10): 2568-75, 1992.
- O'Neill AF, Xia C, Krailo MD, et al.: α-Fetoprotein as a predictor of outcome for children with germ cell tumors: A report from the Malignant Germ Cell International Consortium. Cancer 125 (20): 3649-3656, 2019.
- Dieckmann KP, Radtke A, Geczi L, et al.: Serum Levels of MicroRNA-371a-3p (M371 Test) as a New Biomarker of Testicular Germ Cell Tumors: Results of a Prospective Multicentric Study. J Clin Oncol 37 (16): 1412-1423, 2019.
- Rogers PC, Olson TA, Cullen JW, et al.: Treatment of children and adolescents with stage II testicular and stages I and II ovarian malignant germ cell tumors: A Pediatric Intergroup Study--Pediatric Oncology Group 9048 and Children's Cancer Group 8891. J Clin Oncol 22 (17): 3563-9, 2004.
- Cushing B, Giller R, Cullen JW, et al.: Randomized comparison of combination chemotherapy with etoposide, bleomycin, and either high-dose or standard-dose cisplatin in children and adolescents with high-risk malignant germ cell tumors: a pediatric intergroup study--Pediatric Oncology Group 9049 and Children's Cancer Group 8882. J Clin Oncol 22 (13): 2691-700, 2004.
- Göbel U, Schneider DT, Calaminus G, et al.: Multimodal treatment of malignant sacrococcygeal germ cell tumors: a prospective analysis of 66 patients of the German cooperative protocols MAKEI 83/86 and 89. J Clin Oncol 19 (7): 1943-50, 2001.
- Frazier AL, Hale JP, Rodriguez-Galindo C, et al.: Revised risk classification for pediatric extracranial germ cell tumors based on 25 years of clinical trial data from the United Kingdom and United States. J Clin Oncol 33 (2): 195-201, 2015.
- International Germ Cell Consensus Classification: a prognostic factor-based staging system for metastatic germ cell cancers. International Germ Cell Cancer Collaborative Group. J Clin Oncol 15 (2): 594-603, 1997.
- Shaikh F, Stark D, Fonseca A, et al.: Outcomes of adolescent males with extracranial metastatic germ cell tumors: A report from the Malignant Germ Cell Tumor International Consortium. Cancer 127 (2): 193-202, 2021.
- Motzer RJ, Nichols CJ, Margolin KA, et al.: Phase III randomized trial of conventional-dose chemotherapy with or without high-dose chemotherapy and autologous hematopoietic stem-cell rescue as first-line treatment for patients with poor-prognosis metastatic germ cell tumors. J Clin Oncol 25 (3): 247-56, 2007.
- Fizazi K, Pagliaro L, Laplanche A, et al.: Personalised chemotherapy based on tumour marker decline in poor prognosis germ-cell tumours (GETUG 13): a phase 3, multicentre, randomised trial. Lancet Oncol 15 (13): 1442-50, 2014.
- Thorup J, McLachlan R, Cortes D, et al.: What is new in cryptorchidism and hypospadias--a critical review on the testicular dysgenesis hypothesis. J Pediatr Surg 45 (10): 2074-86, 2010.
- Duhil de Bénazé G, Pacquement H, Faure-Conter C, et al.: Paediatric dysgerminoma: Results of three consecutive French germ cell tumours clinical studies (TGM-85/90/95) with late effects study. Eur J Cancer 91: 30-37, 2018.
- Fonseca A, Xia C, Lorenzo AJ, et al.: Detection of Relapse by Tumor Markers Versus Imaging in Children and Adolescents With Nongerminomatous Malignant Germ Cell Tumors: A Report From the Children's Oncology Group. J Clin Oncol 37 (5): 396-402, 2019.
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed September 5, 2024.