




General Information About Childhood Central Nervous System (CNS) Germ Cell Tumors
Primary brain tumors, including germ cell tumors (GCTs), are a diverse group of diseases that together constitute the most common solid tumors of childhood. The most recent World Health Organization (WHO) Classification of Central Nervous System Tumours implements some molecular parameters, in addition to histology, to define brain tumor entities.[1,2] Some CNS tumor types, such as embryonal tumors and gliomas, are organized according to molecular characterization. However, this updated classification schema does not yet categorize intracranial GCTs using molecular parameters. Tumor location, extent of disease (brain invasion and tumor spread), and type of CNS GCT histology remain important factors that affect treatment and prognosis.
CNS GCTs are broadly classified as germinomatous (commonly referred to as germinoma) and nongerminomatous germ cell tumors (NGGCTs) on the basis of clinicopathological and laboratory features, including tumor markers.[2,3] An alternative therapeutic classification in Japan distinguishes three groups on the basis of their prognosis: good prognosis (e.g., germinoma), intermediate prognosis (e.g., immature teratoma with malignant transformation), and poor prognosis (e.g., yolk sac tumor, choriocarcinoma, embryonal carcinoma, and mixed tumors of those entities).[3]
The PDQ childhood brain tumor treatment summaries are organized primarily according to the WHO Classification of Central Nervous System Tumours.[1,2,3] For a full description of the classification of CNS tumors and a link to the corresponding treatment summary for each type of brain tumor, see the Childhood Brain and Spinal Cord Tumors Summary Index.
Incidence
In Western countries, GCTs represent 3% to 4% of primary brain tumors in children, with a peak incidence from age 10 to 19 years.[4,5] In Japan and other Asian countries, a series reported the incidence of CNS GCTs to be approximately 15% of all pediatric CNS tumors.[5,6,7,8,9] The genetic or environmental reasons for these differences remain unknown.
Overall, males have a higher incidence of GCTs than females. Male patients have a preponderance of pineal-region primary tumors.[10,11] However, male predominance is not noted in patients aged 10 years or younger at the time of diagnosis.[12]
Anatomy
CNS GCTs usually arise in the pineal and/or suprasellar regions of the brain as solitary or multiple lesions (see Figure 1). The most common site of origin is the pineal region (45%), and the second most common site is the suprasellar region (30%) within the infundibulum or pituitary stalk. Both of these sites are considered extra-axial or nonparenchymal CNS locations. Approximately 5% to 10% of patients present with synchronous tumors arising in both the suprasellar and pineal locations. Germinoma is the most frequently observed histology.[8] Other sites that may be involved include the basal ganglia, thalamus, and, less frequently, the ventricles, cerebral hemispheres, and brain stem.[10,11,13] Suprasellar tumors are most common in younger patients, whereas pineal or bifocal presentation predominates in older patients.[12]
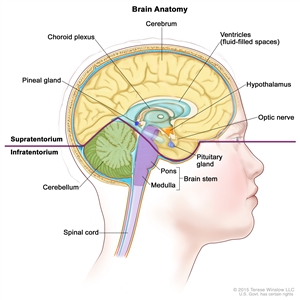
Figure 1. Anatomy of the inside of the brain. The supratentorium contains the cerebrum, ventricles (with cerebrospinal fluid shown in blue), choroid plexus, hypothalamus, pineal gland, pituitary gland, and optic nerve. The infratentorium contains the cerebellum and brain stem.
Clinical Features
The signs and symptoms of CNS GCTs depend on the location of the tumor in the brain, as follows:
- Suprasellar region. Patients with tumors arising in the suprasellar region often present with subtle or overt hormonal deficiencies and may experience a protracted prodrome lasting months to years. Diabetes insipidus caused by antidiuretic hormone deficiency occurs in 70% to 90% of patients and is the most common sentinel symptom. Patients can usually compensate for this deficiency by drinking excessive amounts of fluid for months to years. Eventually, other hormonal symptoms and visual deficits may emerge as the tumor expands dorsally and compresses or invades the optic chiasm and/or fills the third ventricle to cause hydrocephalus.[14,15,16]
- Pineal region. Patients with tumors in the pineal region usually have a shorter history of symptoms than patients with tumors of the suprasellar or basal ganglionic region, with weeks to months of symptoms that include raised intracranial pressure and diplopia related to tectal and aqueductal compression. Signs and symptoms unique to masses in the pineal and posterior third ventricular region include Parinaud syndrome (vertical gaze impairment, convergence nystagmus, and light-near pupillary response dissociation), headache, and nausea and vomiting.
- Bifocal tumors. Patients with bifocal primary tumors present with metasynchronous lesions in the suprasellar and pineal regions.[15] The secondary lesion is often asymptomatic and found on magnetic resonance imaging (MRI). In children with pineal primary tumors, the suprasellar lesion may also be associated with unexplained precocious puberty.
Nonspecific symptoms such as enuresis, anorexia, and psychiatric complaints [17] can lead to delays in a diagnosis. However, signs of increased intracranial pressure or visual changes tend to result in an earlier diagnosis.[18]
Diagnostic Evaluation and Prognostic Factors
Radiographic characteristics of CNS GCTs cannot reliably differentiate germinomas from NGGCTs or other CNS tumors. The diagnosis of GCTs is based on the following:
- Characteristic clinical signs and symptoms supported by neuroimaging.
- GCT marker analysis in the serum and lumbar cerebrospinal fluid (CSF).
- Histology, if necessary.
The diagnosis of a suspected CNS GCT and an assessment of the clinical deficits and extent of metastases can usually be confirmed with the following tests:
- MRI of brain and spine with and without gadolinium.
- Alpha-fetoprotein (AFP) and beta subunit human chorionic gonadotropin (beta-HCG) levels in both serum and CSF, and cytology, if needed. If preoperative CSF can be obtained safely and tumor markers are found to be elevated, histological confirmation may not be needed. Before definitive therapy is initiated, a lumbar CSF assessment for cytology and tumor markers should be performed, if safe, to reconfirm the diagnosis and help monitor treatment response and control. The diagnostic utility of lumbar CSF is better validated and more reliable than that obtained from the ventricles (see Table 1).[18,19]
- Evaluation of pituitary/hypothalamic function.
- Visual-field and acuity examinations for suprasellar or hypothalamic tumors.
If possible, a baseline neuropsychological examination should be performed after symptoms of endocrine deficiency and raised intracranial pressure are resolved.
CNS GCTs can be diagnosed and classified on the basis of histology alone, tumor markers alone, or a combination of both.[19,20,21] A diagnosis of GCTs often requires a tumor biopsy, except when imaging characteristics are present and increased tumor markers (usually AFP and beta-HCG) are found in the serum and/or CSF. The tumor markers AFP and beta-HCG are the most useful, although other markers, such as placental alkaline phosphatase and c-kit, are being investigated (see Table 1). When the tumor markers are negative or mildly elevated but below diagnostic criteria, or if there are any atypical findings, an endoscopic or open biopsy is needed to make a definitive diagnosis.
Distinguishing between different GCT types by CSF protein marker levels alone is somewhat arbitrary, and standards vary across continents. Patients with pure germinomas and teratomas usually present with negative markers, but low levels of beta-HCG can be detected in patients with germinomas.[22] Current international efforts are directed at determining a marker threshold for beta-HCG–secreting germinomas because data suggest that the beta-HCG levels that are used to distinguish germinomas from NGGCTs (50 IU/L in Europe and 100 IU/L in North America) are questionable.
The use of tumor markers and histology in GCT clinical trials is evolving. For example, in the COG-ACNS1123 (NCT01602666) trial, patients were eligible for assignment to the germinoma regimen without biopsy confirmation if they had one of the following:
- Either pineal region tumors or suprasellar primary tumors, normal AFP levels, and beta-HCG levels between 5 and 50 IU/L in serum and/or CSF.
- Bifocal (pineal and suprasellar) involvement or pineal lesions with diabetes insipidus, normal AFP levels, and beta-HCG levels of 100 IU/L or lower in serum and/or CSF.
| Tumor Type | Beta-HCG | AFP | PLAP | c-kit | |
|---|---|---|---|---|---|
| AFP = alpha-fetoprotein; HCG = human chorionic gonadotropin; PLAP = placental alkaline phosphatase; + = positive; +++ = highly positive (elevated); - = negative; ± = equivocal. | |||||
| Germinoma | ± | - | ± | + | |
| Germinoma (syncytiotrophoblastic) | + | - | ± | + | |
| Embryonal carcinoma | ± | + | ± | - | |
| Yolk sac tumor | - | +++ | ± | - | |
| Choriocarcinoma | +++ | - | ± | - | |
| Teratoma | |||||
| Immature teratoma | ± | ± | - | ± | |
| Immature teratoma with malignant components | ± | + | + | ± | |
| Mature teratoma | - | - | - | - | |
| Mixed germ cell tumor | ± | ± | ± | ± | |
There is also an effort to use tumor markers to determine prognosis on the basis of the presence and degree of elevation of AFP and beta-HCG. This is an evolving process, and cooperative groups in North America, Europe, and Japan have adopted slightly different criteria.[23]
Alternative classification schemes for CNS GCTs have been proposed by groups such as the Japanese Pediatric Brain Tumor Study Group for CNS GCTs. This group based their stratification on the prognostic grouping of the differing histological variants, as shown in Table 2.[9]
| Prognostic Group | Tumor Type |
|---|---|
| Good | Germinoma, pure |
| Mature teratoma | |
| Intermediate | Germinoma with syncytiotrophoblastic giant cells |
| Immature teratoma | |
| Mixed tumors mainly composed of germinoma or teratoma | |
| Teratoma with malignant transformation | |
| Poor | Choriocarcinoma |
| Embryonal carcinoma | |
| Mixed tumors mainly composed of choriocarcinoma, yolk sac tumor, or embryonal carcinoma | |
| Yolk sac tumor |
It is crucial that appropriate staging is determined and that germinomas are distinguished from NGGCTs. Chemotherapy and radiation treatment plans differ significantly depending on GCT category and extent of disease.
Cellular and Molecular Classification
The pathogenesis of intracranial GCTs is unknown. The germ cell theory proposes that CNS GCTs arise from primordial germ cells that have aberrantly migrated and undergone malignant transformation. A genome-wide methylation profiling study of 61 GCTs supports this hypothesis.[24] Previous molecular studies that compared the genomic alterations in GCTs showed similar copy-number alterations in both CNS GCTs and systemic GCTs.[25]
An alternative hypothesis, the embryonic cell theory, proposes that GCTs arise from a pluripotent embryonic cell that escapes normal developmental signals and progresses to CNS GCTs.[26,27]
The WHO has classified CNS GCTs into the following groups:[1,2]
- Germinoma.
- Nongerminomatous GCTs.
- Embryonal carcinoma.
- Yolk sac tumor.
- Choriocarcinoma.
- Teratoma.
- Mature teratoma.
- Immature teratoma.
- Teratoma with somatic-type malignancy.
- Mixed GCT.
NGGCTs can consist of one malignant NGGCT type or contain multiple elements of GCT components, including teratomatous or germinomatous constituents.
Recurrent variants in KIT, genes in the MAPK pathway, and genes in the PI3K/mTOR signaling pathway have been identified in CNS GCTs.[28,29,30]
In a retrospective analysis of 82 children and adults with CNS GCTs, chromosome 12p gain was the most frequent copy number alteration. 12p gain was more frequent in NGGCTs (20 of 40, 50%) than germinomas (5 of 42, 12%). 12p gain was associated with worse survival in patients with NGGCTs (10-year overall survival rate, 47% for patients with 12p gain vs. 90% without; P = .02).[31]
Global hypomethylation that mirrors primordial germ cells in early development has also been observed in CNS GCTs.[30]
In an evaluation of 21 cases of CNS germinomas diagnosed between 2000 and 2016, programmed death-ligand 1 (PD-L1) and programmed cell death-1 (PD-1) expression was assessed by immunohistochemistry. Ninety percent of germinomas had germ cell components that stained positively for PD-L1. In addition, tumor-associated lymphocytes stained positive for PD-L1 in more than 75% of cases.[32]
References:
- Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
- WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
- Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016.
- Matsutani M, Sano K, Takakura K, et al.: Primary intracranial germ cell tumors: a clinical analysis of 153 histologically verified cases. J Neurosurg 86 (3): 446-55, 1997.
- Ostrom QT, Gittleman H, Liao P, et al.: CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol 19 (suppl_5): v1-v88, 2017.
- Committee of Brain Tumor Registry of Japan: Report of Brain Tumor Registry of Japan (1969-1996). Neurol Med Chir (Tokyo) 43 (Suppl): i-vii, 1-111, 2003.
- The Committee of Brain Tumor Registry of Japan: Brain Tumor Registry of Japan (2001–2004). Neurol Med Chir (Tokyo) 54 (Suppl): 1-102, 2014. Also available online. Last accessed August 21, 2023.
- Weksberg DC, Shibamoto Y, Paulino AC: Bifocal intracranial germinoma: a retrospective analysis of treatment outcomes in 20 patients and review of the literature. Int J Radiat Oncol Biol Phys 82 (4): 1341-51, 2012.
- Matsutani M; Japanese Pediatric Brain Tumor Study Group: Combined chemotherapy and radiation therapy for CNS germ cell tumors--the Japanese experience. J Neurooncol 54 (3): 311-6, 2001.
- Goodwin TL, Sainani K, Fisher PG: Incidence patterns of central nervous system germ cell tumors: a SEER Study. J Pediatr Hematol Oncol 31 (8): 541-4, 2009.
- Villano JL, Propp JM, Porter KR, et al.: Malignant pineal germ-cell tumors: an analysis of cases from three tumor registries. Neuro Oncol 10 (2): 121-30, 2008.
- Koh KN, Wong RX, Lee DE, et al.: Outcomes of intracranial germinoma-A retrospective multinational Asian study on effect of clinical presentation and differential treatment strategies. Neuro Oncol 24 (8): 1389-1399, 2022.
- Graham RT, Abu-Arja MH, Stanek JR, et al.: Multi-institutional analysis of treatment modalities in basal ganglia and thalamic germinoma. Pediatr Blood Cancer 68 (10): e29172, 2021.
- Kilday JP, Laughlin S, Urbach S, et al.: Diabetes insipidus in pediatric germinomas of the suprasellar region: characteristic features and significance of the pituitary bright spot. J Neurooncol 121 (1): 167-75, 2015.
- Hoffman HJ, Otsubo H, Hendrick EB, et al.: Intracranial germ-cell tumors in children. J Neurosurg 74 (4): 545-51, 1991.
- Sethi RV, Marino R, Niemierko A, et al.: Delayed diagnosis in children with intracranial germ cell tumors. J Pediatr 163 (5): 1448-53, 2013.
- Malbari F, Gershon TR, Garvin JH, et al.: Psychiatric manifestations as initial presentation for pediatric CNS germ cell tumors, a case series. Childs Nerv Syst 32 (8): 1359-62, 2016.
- Crawford JR, Santi MR, Vezina G, et al.: CNS germ cell tumor (CNSGCT) of childhood: presentation and delayed diagnosis. Neurology 68 (20): 1668-73, 2007.
- Allen J, Chacko J, Donahue B, et al.: Diagnostic sensitivity of serum and lumbar CSF bHCG in newly diagnosed CNS germinoma. Pediatr Blood Cancer 59 (7): 1180-2, 2012.
- Rosenblum MK, Nakazato Y, Matsutani M: Germ cell tumours. In: Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016, pp 286-91.
- Murray MJ, Bartels U, Nishikawa R, et al.: Consensus on the management of intracranial germ-cell tumours. Lancet Oncol 16 (9): e470-e477, 2015.
- Frazier AL, Olson TA, Schneider DT, et al.: Germ cell tumors. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Lippincott Williams and Wilkins, 2015, pp 899-918.
- Calaminus G, Bamberg M, Harms D, et al.: AFP/beta-HCG secreting CNS germ cell tumors: long-term outcome with respect to initial symptoms and primary tumor resection. Results of the cooperative trial MAKEI 89. Neuropediatrics 36 (2): 71-7, 2005.
- Fukushima S, Yamashita S, Kobayashi H, et al.: Genome-wide methylation profiles in primary intracranial germ cell tumors indicate a primordial germ cell origin for germinomas. Acta Neuropathol 133 (3): 445-462, 2017.
- Schneider DT, Zahn S, Sievers S, et al.: Molecular genetic analysis of central nervous system germ cell tumors with comparative genomic hybridization. Mod Pathol 19 (6): 864-73, 2006.
- Sano K, Matsutani M, Seto T: So-called intracranial germ cell tumours: personal experiences and a theory of their pathogenesis. Neurol Res 11 (2): 118-26, 1989.
- Teilum G: Embryology of ovary, testis, and genital ducts. In: Teilum G: Special Tumors of Ovary and Testis and Related Extragonadal Lesions: Comparative Pathology and Histological Identification. J. B. Lippincott, 1976, pp 15-30.
- Wang L, Yamaguchi S, Burstein MD, et al.: Novel somatic and germline mutations in intracranial germ cell tumours. Nature 511 (7508): 241-5, 2014.
- Takami H, Fukuoka K, Fukushima S, et al.: Integrated clinical, histopathological, and molecular data analysis of 190 central nervous system germ cell tumors from the iGCT Consortium. Neuro Oncol 21 (12): 1565-1577, 2019.
- Schulte SL, Waha A, Steiger B, et al.: CNS germinomas are characterized by global demethylation, chromosomal instability and mutational activation of the Kit-, Ras/Raf/Erk- and Akt-pathways. Oncotarget 7 (34): 55026-55042, 2016.
- Satomi K, Takami H, Fukushima S, et al.: 12p gain is predominantly observed in non-germinomatous germ cell tumors and identifies an unfavorable subgroup of central nervous system germ cell tumors. Neuro Oncol 24 (5): 834-846, 2022.
- Wildeman ME, Shepard MJ, Oldfield EH, et al.: Central Nervous System Germinomas Express Programmed Death Ligand 1. J Neuropathol Exp Neurol 77 (4): 312-316, 2018.