




General Information About Childhood Astrocytomas, Other Gliomas, and Glioneuronal / Neuronal Tumors
Primary brain tumors, including gliomas, are a diverse group of diseases that together constitute the most common solid tumors of childhood. Brain tumors are classified according to histology and molecular features, but tumor location and extent of spread are also important factors that affect treatment and prognosis. Histological features, immunohistochemical analysis, and cytogenetic and molecular genetic findings are used in tumor diagnosis and classification.
Gliomas are thought to arise from neural stem and progenitor cells that are present in the brain and spinal cord. Gliomas are classified on the basis of histological and molecular features, and they represent the most common type of central nervous system (CNS) tumor in children.
Historically, pediatric gliomas were classified into low-grade (World Health Organization [WHO] grades 1–2) and high-grade (WHO grades 3–4) gliomas on the basis of histological features. However, the incorporation of molecular biomarkers has led to a new classification scheme. According to the 2021 WHO Classification of Tumours: Central Nervous System Tumours (5th edition), gliomas, glioneuronal tumors, and neuronal tumors are broadly classified into adult-type diffuse gliomas, pediatric-type diffuse low-grade gliomas, pediatric-type diffuse high-grade gliomas, circumscribed astrocytic gliomas, glioneuronal and neuronal tumors, and ependymal tumors.[1,2] Within these tumor types, various subtypes are recognized, and histological grading ranging from grade 1 to grade 4 is applied to some. Most children with circumscribed astrocytic gliomas, pediatric-type diffuse low-grade gliomas, and glioneuronal and neuronal tumors have a relatively favorable prognosis, especially when a complete surgical resection can be accomplished. Children with pediatric-type diffuse high-grade gliomas generally have a poor prognosis. For information about ependymal tumors, see Childhood Ependymoma Treatment.
The PDQ childhood brain tumor treatment summaries are organized primarily according to the 2021 WHO CNS classification.[1,2]
Anatomy
Childhood gliomas can occur anywhere in the CNS (see Figure 1). For the most common CNS location for each tumor type, see Table 2. 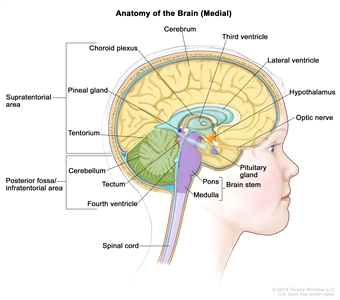
Figure 1. Anatomy of the inside of the brain, showing the cerebrum, cerebellum, brain stem, spinal cord, optic nerve, hypothalamus, and other parts of the brain.
Clinical Features
Presenting symptoms for childhood gliomas depend on the following:
- Anatomical location.
- Size of the tumor.
- Rate of tumor growth.
- Chronological and developmental age of the child.
Infants and young children with circumscribed gliomas (most commonly pilocytic astrocytomas) and, less frequently, diffuse astrocytomas, involving the hypothalamus may present with diencephalic syndrome, which is manifested by failure to thrive in an emaciated, seemingly euphoric child. Such children may have little in the way of other neurological findings, but may present with macrocephaly, intermittent lethargy, and/or visual impairment.[3]
Children with diffuse midline gliomas centered in the pons (previously called diffuse intrinsic pontine gliomas [DIPGs]) may present with the following classic triad of symptoms; however, children may present with only one or two of these symptoms at diagnosis:
- Cranial neuropathies, particularly abducens paresis.
- Long tract signs.
- Ataxia.
Obstructive hydrocephalus caused by expansion of the pons can also be a presenting symptom. Nonspecific symptoms may also occur, including behavioral changes and decreased school performance.
The presentation of circumscribed astrocytomas (e.g., pilocytic astrocytomas) in the brain stem depends on the tumor location. Common presenting symptoms include the following:[4]
- Raised intracranial pressure with associated hydrocephalus.
- Unilateral hemiparesis.
- Unilateral cranial neuropathies.
- Ataxia.
Diagnostic Evaluation
The initial diagnostic evaluation of patients with gliomas includes magnetic resonance imaging (MRI) with and without contrast of the brain and/or spine. The risk of neuraxis dissemination is tumor type dependent, and complete neuraxis imaging, including MRIs of the brain and total spine, may be performed in select patients. In most cases, the specific diagnosis is determined after surgical intervention and pathological classification.
Primary tumors of the brain stem are most often diagnosed on the basis of clinical findings and on neuroimaging studies using MRI, as follows:[5]
- Diffuse midline glioma centered in the pons (DIPG). A presumptive diagnosis of DIPG based on classic imaging and clinical features, in the absence of a histological diagnosis, has been routinely employed. Increasingly however, histological confirmation is obtained for both entry into research studies and molecular characterization of the tumor.[6] Given the technical challenges of pontine biopsies, the procedure is best undertaken by an experienced pediatric neurosurgeon to minimize the risk of irreversible neurological complications.[7,8,9,10,11] Biopsy is recommended for pontine tumors when the diagnosis is uncertain based on imaging findings.
- Non-DIPG brain stem tumors. Biopsy or resection is generally indicated for non-DIPG brain stem tumors.
Lumbar punctures examining the cerebrospinal fluid for circulating tumor cells are not commonly performed in children with these tumor types.
WHO Classification of Childhood CNS Astrocytomas, Gliomas, and Glioneuronal/Neuronal Tumors
The pathological classification of pediatric brain tumors is a highly specialized area that continues to evolve. Rapid advances in molecular genetics have led to major improvements in the accurate diagnosis of brain tumors over the past decade. At the same time, many novel brain tumor entities have been recognized on the basis of unique molecular features. Examination of the diagnostic tissue by an experienced neuropathologist is strongly recommended, along with molecular testing, if available.
According to the 2021 WHO CNS classification, gliomas and glioneuronal/neuronal tumors occurring predominantly in childhood are broadly classified as follows:
- Pediatric-type diffuse high-grade gliomas.
- Pediatric-type diffuse low-grade gliomas.
- Circumscribed astrocytic gliomas.
- Glioneuronal and neuronal tumors.
- Ependymal tumors. For more information, see Childhood Ependymoma Treatment.
Within each tumor type, various subtypes are recognized on the basis of histological and molecular features.
The 2021 WHO CNS classification recommends a layered report structure as follows:[1,2]
- Integrated diagnosis (combined tissue-based histological and molecular diagnosis).
- Histological diagnosis.
- CNS WHO grade.
- Molecular information (listed).
WHO CNS tumor grading
Whereas CNS tumors were previously graded on histopathological grounds and clinical behavior alone (clinicopathological grading), the 2021 WHO CNS grading scheme employs combined histological and molecular grading for many tumor types.[1] Histological grading ranges from 1 to 4, but not all grades are applied to all tumor types, and some tumor types are not graded.
The 2021 WHO CNS classification and grading of the most common types/subtypes of gliomas, glioneuronal tumors, and neuronal tumors (excluding ependymal tumors) occurring in childhood and adolescence are shown in Table 1.
| Tumor Type/Subtype | WHO CNS Grades | |
|---|---|---|
| Pediatric-type diffuse high-grade gliomas: | ||
| Diffuse midline glioma, H3 K27-altered | 4 | |
| Diffuse pediatric-type high-grade glioma, H3-wild type and IDH-wild type | 4 | |
| Infant-type hemispheric glioma | Not assigned | |
| Pediatric-type diffuse low-grade gliomas: | ||
| Diffuse low-grade glioma, MAPK pathway-altered | Not assigned | |
| Diffuse astrocytoma,MYB- orMYBL1-altered | 1 | |
| Circumscribed astrocytic gliomas: | ||
| Pilocytic astrocytoma | 1 | |
| High-grade astrocytoma with piloid features | Not assigned | |
| Pleomorphic xanthoastrocytoma | 2, 3 | |
| Subependymal giant cell astrocytoma | 1 | |
| Glioneuronal and neuronal tumors: | ||
| Ganglioglioma | 1 | |
| Desmoplastic infantile ganglioglioma/desmoplastic infantile astrocytoma | 1 | |
| Dysembryoplastic neuroepithelial tumor | 1 | |
CNS location
Childhood gliomas can occur anywhere in the CNS, although each tumor type tends to occur in specific anatomical locations (see Table 2).
| Tumor Type | Common CNS Location |
|---|---|
| Circumscribed astrocytic gliomas | Cerebellum, optic nerve, optic chiasm/hypothalamus, thalamus and basal ganglia, brain stem, cerebral hemispheres, and spinal cord (rare) |
| Ganglioglioma | Cerebrum, brain stem; occasionally other locations |
| Diffuse midline glioma, H3 K27-altered | Pons, thalamus, spinal cord, and other midline structures |
| Diffuse pediatric-type high-grade glioma, H3-wild type and IDH-wild type | Cerebrum; occasionally other locations |
Cerebellum: More than 80% of gliomas located in the cerebellum are pilocytic astrocytomas (WHO grade 1) and often cystic; most of the remainder represent pediatric-type diffuse low-grade gliomas.[12] High-grade gliomas in the cerebellum are rare.
Brain stem: The term brain stem glioma is a generic description that refers to any tumor of glial origin arising in the brain stem, inclusive of the midbrain, pons, and medulla. While other histologies (e.g., ganglioglioma) can occur in the brain stem, the following two histologies predominate:
- Diffuse midline glioma, H3 K27-altered, which are centered in the pons.[13] These were commonly referred to as diffuse intrinsic pontine gliomas (DIPG) due to their anatomical location. For more information about diffuse midline glioma, H3 K27-altered, see the Genomics of Gliomas, Glioneuronal Tumors, and Neuronal tumors section.
- Pilocytic astrocytomas, which occur throughout the brain stem.
Tumors with exophytic components are overwhelmingly pilocytic astrocytomas.[14] DIPG accounts for approximately 75% to 80% of pediatric brain stem tumors.[15] Most children with DIPGs are diagnosed between the ages of 5 and 10 years. Focal pilocytic astrocytomas in the brain stem occur less frequently.[4]
Optic pathway and hypothalamus: Most tumors arising within the optic pathway (i.e., optic nerve, chiasm, and optic radiations) represent pilocytic astrocytomas, and rarely pediatric-type diffuse low-grade gliomas.[12]
Cerebrum: Most tumors arising in the cerebral hemispheres comprise circumscribed astrocytic gliomas and pediatric-type diffuse low-grade gliomas, followed by pediatric-type diffuse high-grade gliomas.[12]
Genomics of Gliomas, Glioneuronal Tumors, and Neuronal Tumors
Selected cancer susceptibility syndromes associated with pediatric glioma
Neurofibromatosis type 1 (NF1)
Children with NF1 have an increased propensity to develop low-grade gliomas, especially in the optic pathway. Up to 20% of patients with NF1 will develop an optic pathway glioma. Most children with NF1-associated optic nerve gliomas are asymptomatic and/or have nonprogressive symptoms and do not require antitumor treatment. Screening magnetic resonance imaging (MRI) in asymptomatic patients with NF1 is usually not indicated, although some investigators perform baseline MRI for young children who cannot undergo detailed ophthalmologic examinations.[16]
The diagnosis is often based on compatible clinical findings and imaging features. Histological confirmation is rarely needed at the time of diagnosis. When biopsies are performed, these tumors are predominantly pilocytic astrocytomas.[12]
Indications for treatment vary, and are often based on the goal of preserving vision.
Very rarely, patients with NF1 develop high-grade gliomas. Sometimes, this tumor is the result of a transformation of a lower-grade tumor.[17]
Tuberous sclerosis
Patients with tuberous sclerosis have a predilection for developing subependymal giant cell astrocytoma (SEGA). Variants in either TSC1 or TSC2 cause constitutive activation of the mammalian target of rapamycin complex 1 (mTORC1) signaling pathway, leading to increases in proliferation. SEGAs are responsive to molecularly targeted approaches with mTORC1 pathway inhibitors.[18][Level of evidence C2] Patients with tuberous sclerosis are also at risk of developing cortical tubers and subependymal nodules.
Molecular features and recurrent genomic alterations
Recurrent genomic alterations resulting in constitutive activation of the mitogen-activated protein kinase (MAPK) pathway, most commonly involving the BRAF gene, represent the primary (and often sole) oncogenic driver in the vast majority of pediatric low-grade gliomas, including pilocytic/pilomyxoid astrocytomas, gangliogliomas, and others.[12] As a result, most of these tumors are amenable to molecular targeted therapies.
More complex tumor genomes are characteristic of pediatric diffuse high-grade gliomas. These complex genomes include recurrent genomic alterations in the H3 histone encoding genes (e.g., H3F3A, HIST1H3B), DNA damage repair pathways (e.g., TP53, PPM1D, ATM, MDM2), chromatin modifiers (e.g., ATRX, BCOR, SETD2), cell cycle pathways (e.g., CDKN2A, CDKN2B, RB1), and/or oncogene amplifications (PDGFR, VEGFR2, KIT, MYC, MYCN).[19] For most of these tumors, existing conventional and molecular targeted therapies have limited efficacy.
A rare subset of pediatric high-grade gliomas arising in patients with inheritable biallelic mismatch repair deficiency (bMMRD) is characterized by an extraordinarily high mutational burden. Correctly identifying these patients at the time of diagnosis is critical because of intrinsic resistance to temozolomide and responsiveness to treatment with immune checkpoint inhibitors.[20][Level of evidence C3]; [21]
BRAF::KIAA1549
BRAF activation in pilocytic astrocytoma occurs most commonly through a BRAF::KIAA1549 gene fusion, resulting in a fusion protein that lacks the BRAF autoregulatory domain.[22] This fusion is seen in most infratentorial and midline pilocytic astrocytomas, but is present at lower frequency in supratentorial (hemispheric) tumors.[12]
Presence of the BRAF::KIAA1549 fusion is associated with improved clinical outcome (progression-free survival [PFS] and overall survival [OS]) in patients with pilocytic astrocytoma.[23]; [24][Level of evidence C1] Progression to high-grade gliomas is very rare for pediatric gliomas with the BRAF::KIAA1549 fusion.[24]
BRAFvariants
Activating point variants in BRAF, most commonly BRAF V600E, are present in a subset of pediatric gliomas and glioneuronal tumors across a wide spectrum of histologies, including pleomorphic xanthoastrocytoma, pilocytic astrocytoma, ganglioglioma, desmoplastic infantile ganglioglioma/astrocytoma, and others.[12] Some low-grade, infiltrative, pediatric gliomas with an alteration in a MAPK pathway gene, including BRAF, and often resembling diffuse low-grade astrocytoma or oligodendroglioma histologically, are now classified as diffuse low-grade glioma, MAPK pathway altered.[1,25]
Retrospective clinical studies have shown the following:
- In a retrospective series of more than 400 children with low-grade gliomas, 17% of tumors had BRAF V600E variants. The 10-year PFS rate was 27% for patients with BRAF V600E variants, compared with 60% for patients whose tumors did not harbor that variant. Additional factors associated with this poor prognosis included subtotal resection and CDKN2A deletion.[26][Level of evidence C2] Even in patients who underwent a gross-total resection, recurrence was noted in one-third, suggesting that BRAF V600E tumors have a more invasive phenotype than do other low-grade glioma variants.
- In a similar analysis, children with diencephalic low-grade astrocytomas with a BRAF V600E variant had a 5-year PFS rate of 22%, compared with a PFS rate of 52% in children with wild-type BRAF.[27][Level of evidence C2]
- The frequency of the BRAF V600E variant was significantly higher in pediatric low-grade gliomas that transformed to high-grade gliomas (8 of 18 patients) than was the frequency of the variant in tumors that did not transform to high-grade gliomas (10 of 167 cases).[24]
NF1variants
Somatic alterations in NF1 are seen most frequently in children with NF1 and are associated with germline alterations in the tumor suppressor NF1. Loss of heterozygosity for NF1 represents the most common somatic alteration in these patients followed by inactivating variants in the second NF1 allele, and consistent with a second hit required for tumorigenesis. While most NF1 patients with low-grade gliomas have an excellent long-term prognosis, secondary transformation into high-grade glioma may occur in a small subset. Genomically, transformation is associated with the acquisition of additional oncogenic drivers, such as loss of function alterations in CDKN2A, CDKN2B and/or ATRX. Primary high-grade gliomas may also occur in patients with NF1 but are exceedingly rare. Genomic alterations involving the MAPK signaling pathway other than NF1 are very uncommon in gliomas occurring in children with NF1.[17]
ALK,NTRK1,NTRK2,NTRK3, orROS1gene fusions
High-grade gliomas with distinctive molecular characteristics arise in infants, typically in those diagnosed during the first year of life.[28,29,30] These tumors are characterized by recurrent oncogenic gene fusions involving ALK, NTRK1, NTRK2, NTRK3, or ROS1 as the primary and, typically, sole oncogenic driver. Infants with this type of glioma, now classified as infant-type hemispheric glioma, have a much better prognosis compared with older children with high-grade gliomas. Remarkably, these tumors may evolve from high-grade to low-grade histology over time, and it remains unclear how much this phenomenon is a consequence of natural disease history versus treatment-induced changes.[28]
ROS1 gene fusions have also been reported in gliomas occurring in older children and adults. A retrospective meta-analysis that included 40 children older than 1 year revealed that ROS1 gene fusions occurred in diverse glioma histologies, including diffuse high-grade and low-grade gliomas and glioneuronal tumors.[30] Similar to ROS1-altered cases occurring in infants, tumor variants in other known driver genes were rare. However, tumor copy number alterations were more frequent in older children than infants.
Other genomic alterations
As an alternative to BRAF activation or NF1 loss, other primary oncogenic driver alterations along the MAPK signaling pathway have been observed in pilocytic astrocytomas and other pediatric-type gliomas. These include oncogenic variants and/or fusions involving FGFR1, FGFR2, PTPN11, RAF1,NTRK2, and others.[12,31,32]
Low-grade gliomas with rearrangements in the MYB family of transcription factors [12,33,34] have now been classified as a separate entity: diffuse astrocytoma, MYB- or MYBL1-altered, WHO grade 1.[1]
Angiocentric gliomas
Angiocentric gliomas typically arise in children and young adults as cerebral tumors presenting with seizures.[35]
Two reports in 2016 identified MYB gene alterations as being present in almost all cases diagnosed as angiocentric glioma, with QKI being the primary fusion partner in cases where fusion-partner testing was possible.[32,36] While angiocentric gliomas most commonly occur supratentorially, brain stem angiocentric gliomas with MYB::QKI fusions have also been reported.[37,38]
Astroblastomas,MN1-altered
Astroblastomas are defined histologically as glial neoplasms composed of GFAP-positive cells and contain astroblastic pseudorosettes that often demonstrate sclerosis. Astroblastomas are diagnosed primarily in childhood through young adulthood.[35]
The following studies have described genomic alterations associated with astroblastoma:
- A report describing a molecular classification of CNS primitive neuroectodermal tumors (PNETs) identified an entity called CNS high-grade neuroepithelial tumor with MN1 alteration (CNS HGNET-MN1) that was characterized by gene fusions involving MN1.[39] Most tumors with a histological diagnosis of astroblastoma (16 of 23) belonged to this molecularly defined entity.
- A report of 27 histologically defined astroblastomas found that 10 cases had MN1 rearrangements, 7 cases had BRAF rearrangements, and 2 cases had RELA rearrangements.[40] Methylation array analysis showed that the cases with MN1 rearrangements clustered with CNS HGNET-MN1, the BRAF-altered cases clustered with pleomorphic xanthoastrocytomas, and the RELA cases clustered with ependymomas.
- Genomic evaluation of eight cases of astroblastoma identified four with MN1 alterations. Of the remaining four cases, two had genomic alterations consistent with high-grade glioma and two cases could not be classified on the basis of their molecular characteristics.[41]
- One study described eight cases of astroblastoma. All five cases that underwent fluorescence in situ hybridization analysis showed MN1 rearrangements.[42]
These reports suggest that the histological diagnosis of astroblastoma encompasses a heterogeneous group of genomically defined entities. Astroblastomas with MN1 fusions represent a distinctive subset of histologically diagnosed cases.[43]
IDH1andIDH2variants
IDH1- and IDH2-altered tumors occur in the pediatric population as low-grade gliomas (WHO Grade 2), high-grade gliomas (WHO Grade 3 and 4), and oligodendrogliomas with codeletion of 1p and 19q. For more information about IDH1- and IDH2-altered gliomas, see the IDH1 and IDH2 variants section in the Molecular features of pediatric-type high-grade gliomas section.
Molecular features of pediatric-type high-grade gliomas
Pediatric high-grade gliomas are biologically distinct from those arising in adults.[44,45,46,47]
Subgroups identified using DNA methylation patterns
Pediatric-type high-grade gliomas can be separated into distinct subgroups on the basis of epigenetic patterns (DNA methylation). These subgroups show distinguishing chromosome copy number gains/losses and gene variants in the tumor.[19,48,49] Particularly distinctive subtypes of pediatric high-grade gliomas are those with recurring variants at specific amino acids in histone genes, and together these account for approximately one-half of pediatric high-grade gliomas.[19]
The following pediatric-type high-grade glioma subgroups were identified on the basis of their DNA methylation patterns, and they show distinctive molecular and clinical characteristics:[19]
Genomic alterations associated with diffuse midline gliomas
The histone K27 variants: H3.3 (H3F3A) and H3.1 (HIST1H3Band, rarely,HIST1H3C) variants at K27 and EZHIP
The histone K27–altered cases occur predominantly in middle childhood (median age, approximately 10 years), are almost exclusively midline (thalamus, brain stem, and spinal cord), and carry a very poor prognosis. The 2021 WHO classification groups these cancers into a single entity: diffuse midline glioma, H3 K27-altered. However, there are clinical and biological distinctions between cases with H3.3 and H3.1 variants, as described below.[1]
Diffuse midline glioma, H3 K27-altered, is defined by loss of H3 K27 trimethylation either due to an H3 K27M variant or, less commonly, overexpression of EZHIP. This entity includes most high-grade gliomas located in the thalamus, pons (diffuse intrinsic pontine gliomas [DIPGs]), and spinal cord, predominantly in children, but also in adults.[50]
H3.3 K27M: H3.3 K27M cases occur throughout the midline and pons, account for approximately 60% of cases in these locations, and commonly present between the ages of 5 and 10 years.[19] The prognosis for H3.3 K27M patients is especially poor, with a median survival of less than 1 year; the 2-year survival rate is less than 5%.[19] Leptomeningeal dissemination is frequently observed in H3.3 K27M patients.[51]
H3.1 K27M: H3.1 K27M cases are approximately fivefold less common than H3.3 K27M cases. They occur primarily in the pons and present at a younger age than other H3.3 K27M patients (median age, 5 years vs. 6–10 years). These patients have a slightly more favorable prognosis than do H3.3 K27M patients (median survival, 15 months vs. 11 months). Variants in ACVR1, which is also the variant observed in the genetic condition fibrodysplasia ossificans progressiva, are present in a high proportion of H3.1 K27M cases.[19,52,53]
H3.2 K27M: Rarely, K27M variants are also identified in H3.2 (HIST2H3C) cases.[19]
A subset of tumors with H3 K27 variants will have a BRAF V600E or FGFR1 co-variant. A retrospective cohort of 29 tumors combined with 31 cases previously reported in the literature demonstrated a somewhat higher propensity for a thalamic location. These cases exhibit a unique DNA methylation cluster that is distinct from other diffuse midline glioma subgroups and glioma subtypes with BRAF or FGFR1 alterations. The median survival for these patients exceeded 3 years.[54] A separate retrospective study of pediatric and adult patients with H3 K27-altered gliomas revealed BRAF V600E variants in 5.8% (9 of 156) and FGFR1 variants in 10.9% (17 of 156) of patients younger than 20 years.[55] Other recurrent genetic alterations detected in pediatric patients included variants in TP53, ATRX, PIK3CA, and amplifications of PDGFRA and KIT. FGFR1 variants were noted to be more frequent in patients older than 20 years (31.8%, 47 of 148).
EZHIP overexpression: The small minority of patients with diffuse midline gliomas lacking histone H3 variants often show EZHIP overexpression.[50] EZHIP inhibits PRC2 activity, leading to the same loss of H3 K27 trimethylation that is induced by H3 K27M variants.[56] Overexpression of EZHIP is likewise observed in posterior fossa type A ependymomas, which also shows loss of H3 K27 methylation.[57]
H3.3 (H3F3A) variant at G34
The H3.3 G34 subtype arises from H3.3 glycine 34 to arginine/valine (G34R/V) variants.[48,49] This subtype presents in older children and young adults (median age, 14–18 years) and arises exclusively in the cerebral cortex.[48,49] H3.3 G34 cases commonly have variants in TP53 and ATRX (95% and 84% of cases, respectively, in one large series) and show widespread hypomethylation across the whole genome. In a series of 95 patients with the H3.3 G34 subtype, 44% of patients also had a variant in PDGFRA at the time of diagnosis, and 81% of patients had PDGFRA variants observed at relapse.[58]
Patients with H3F3A variants are at high risk of treatment failure,[59] but the prognosis is not as poor as that of patients with histone 3.1 or 3.3 K27M variants.[49] O-6-methylguanine-DNA methyltransferase (MGMT) methylation is observed in approximately two-thirds of cases, and aside from the IDH1-altered subtype (see below), the H3.3 G34 subtype is the only pediatric high-grade glioma subtype that demonstrates MGMT methylation rates exceeding 20%.[19]
IDH1andIDH2variants
IDH1- and IDH2-altered tumors occur in the pediatric population as low-grade gliomas (WHO grade 2), high-grade gliomas (WHO grades 3 and 4), and oligodendrogliomas with codeletion of 1p and 19q.[60]
- IDH1 variants are much more common than IDH2 variants, accounting for approximately 90% of pediatric IDH-altered CNS tumors.
- IDH-altered low-grade gliomas are more common than IDH-altered high-grade gliomas, accounting for approximately three-fourths of IDH-altered pediatric glioma cases.
- Oligodendrogliomas with IDH variants represent approximately 20% of pediatric CNS tumors with IDH variants.
- The median age at diagnosis for pediatric patients with IDH-altered tumors is approximately 16 years, and IDH-altered CNS tumors are very uncommon in children aged 10 years and younger.
- Like astrocytomas with IDH variants in adults, those in affected children commonly have TP53 variants (approximately 90% of cases) and ATRX variants (approximately 50%).
- Like IDH-altered, low-grade gliomas in adults, low-grade tumors in pediatric patients can also show progression to high-grade gliomas.
IDH1-altered cases represent a small percentage of high-grade gliomas (approximately 5%–10%) seen in pediatrics, and are almost exclusively older adolescents (median age in a pediatric population, 16 years) with hemispheric tumors.[19,60] These tumors are classified under adult-type diffuse glioma, as astrocytoma, IDH-altered in the 2021 WHO CNS classification. IDH1-altered cases often show TP53 variants, MGMT promoter methylation, and a glioma-CpG island methylator phenotype (G-CIMP).[48,49]
Pediatric patients with IDH1 variants have a more favorable prognosis than patients with other types of high-grade gliomas.[19] A retrospective multi-institutional review of pediatric patients with IDH-altered gliomas and available outcome data (n = 76) reported a 5-year PFS rate of 44% (95% CI, 25%–59%) and a 5-year OS rate of 92% (95% CI, 79%–97%).[60] Approximately 25% of the gliomas in the cohort were classified as high grade. There was no difference in 5-year PFS rates observed between tumor grades. However, patients with high-grade tumors had a worse 5-year OS rate of 75% (95% CI, 40%–91%).
Rare, IDH-altered, high-grade gliomas have been reported to occur in children with mismatch repair–deficiency syndromes (Lynch syndrome or constitutional mismatch repair deficiency syndrome).[61] These tumors, termed primary mismatch repair–deficient IDH-altered astrocytomas (PMMRDIAs), could be distinguished from other IDH-altered gliomas by methylation profiling. PMMRDIAs have molecular features that are distinct from most IDH-altered gliomas, including a hypervariant phenotype and frequent activation of receptor tyrosine kinase pathways. Patients with PMMRDIAs have a markedly worse prognosis than patients with other IDH-altered gliomas, with a median survival of 15 months.
Pleomorphic xanthoastrocytoma (PXA)–like
Approximately 10% of pediatric high-grade gliomas have DNA methylation patterns that are PXA-like.[49] PXA-like cases commonly have BRAF V600E variants and a relatively favorable outcome (approximately 50% survival at 5 years).[19,59]
High-grade astrocytoma with piloid features
This entity was included in the 2016 WHO classification (called pilocytic astrocytoma with anaplasia) to describe tumors with histological features of pilocytic astrocytoma, increased mitotic activity, and additional high-grade features. The current nomenclature was adopted in the 2021 WHO classification. A more recent publication described a cohort of 83 cases with these histological features (referred to as anaplastic astrocytoma with piloid features) that shared a common DNA methylation profile, which is distinct from the methylation profiles of other gliomas. These tumors occurred more often in adults (median age, 41 years), and they harbored frequent deletions of CDKN2A/B, MAPK pathway alterations (most often in the NF1 gene), and variants or deletions of ATRX. They are associated with a clinical course that is intermediate between pilocytic astrocytoma and IDH–wild-type glioblastoma.[62]
Other variants
Pediatric patients with glioblastoma multiforme high-grade glioma whose tumors lack both histone variants and IDH1 variants represent approximately 40% of pediatric glioblastoma multiforme cases.[19,63] This is a heterogeneous group, with higher rates of gene amplifications than other pediatric high-grade glioma subtypes. The most commonly amplified genes are PDGFRA, EGFR, CCND/CDK, and MYC/MYCN.[48,49] MGMT promoter methylation rates are low in this group.[63] One report divided this group into three subtypes. The subtype characterized by high rates of MYCN amplification showed the poorest prognosis, while the subtype characterized by TERT promoter variants and EGFR amplification showed the most favorable prognosis. The third group was characterized by PDGFRA amplification.[63]
High-grade gliomas in infants
Infants and young children with high-grade gliomas appear to have tumors with distinctive molecular characteristics [28,29] when compared with tumors of older children and adults with high-grade gliomas. An indication of this difference was noted with the application of DNA methylation analysis to pediatric high-grade tumors, which found that approximately 7% of pediatric patients with a histological diagnosis of high-grade glioma had tumors with methylation patterns more closely resembling those of low-grade gliomas.[19] Ten of 16 infants (younger than 1 year) with a high-grade glioma diagnosis were in this methylation array–defined group.[19] The 5-year survival rate for patients in this report diagnosed at younger than 1 year exceeded 60%, while the 5-year survival rate for patients aged 1 to 3 years and older was less than 20%.
Two studies of the molecular characteristics of high-grade gliomas in infants and young children have further defined the distinctive nature of tumors arising in children younger than 1 year. A key finding from both studies is the importance of gene fusions involving tyrosine kinases (e.g., ALK, NTRK1, NTRK2, NTRK3, and ROS1) in patients in this age group. Both studies also found that infants with high-grade gliomas whose tumors have these gene fusions have survival rates much higher than those of older children with high-grade gliomas.[28,29]
The first study presented data for 118 children younger than 1 year with a low-grade or high-grade glioma diagnosis who had tumor tissue available for genomic characterization.[28] Approximately 75% of the cases were classified as low grade, but the diminished utility of histological classification in this age group was illustrated by the relatively low OS rate for the low-grade cohort (71%) and the relatively favorable survival for the high-grade cohort (55%). Rates of surgical resection were higher for patients with high-grade tumors, a result of many of the low-grade tumors occurring in midline locations while the high-grade tumors were found in supratentorial locations. This finding may also help to explain the relative outcomes for the two groups. Genomic characterization divided the infant glioma population into the following three groups, the first of which included patients with high-grade gliomas:
- Group 1 tumors were receptor tyrosine kinase driven and primarily high grade (83%). These tumors harbored lesions in ALK, ROS1, NTRK, and MET. The median age at diagnosis was 3 months, and OS rates were approximately 60%.
- Group 2 tumors were RAS/MAPK driven and were all hemispheric low-grade gliomas, representing one-fourth of hemispheric gliomas in infants. BRAF V600E was the most common alteration, followed by FGFR1 alterations and BRAF fusions. This group had a median age at presentation of 8 months and had the most favorable outcome (10-year OS rate, 93%).
- Group 3 tumors were RAS/MAPK driven with low-grade histology and midline presentation (approximately 80% optic pathway/hypothalamic gliomas). Most group 3 tumors showed either BRAF fusions or BRAF V600E. Median age at diagnosis was 7.5 months. The 5-year progression-free survival (PFS) rate was approximately 20%, and the 10-year OS rate was approximately 50% (far inferior to that of optic pathway/hypothalamic gliomas in children aged >1 year).
The second study focused on tumors from children younger than 4 years with a pathological diagnosis of WHO grades 2, 3, and 4 gliomas, astrocytomas, or glioneuronal tumors. Among the 191 tumors studied that met inclusion criteria, 61 had methylation profiles consistent with glioma subtypes that occur in older children (e.g., IDH1, diffuse midline glioma H3 K27-altered, SEGA, pleomorphic xanthoastrocytoma, etc.). The remaining 130 cases were called the intrinsic set and were the focus of additional molecular characterization:[29]
- The intrinsic set contained most of the patients diagnosed before age 1 year (49 of 63 patients, 78%) and had a median age of 7.2 months. Tumors were frequently in a superficial hemispheric location, often involving the meninges, and had a well-defined border with adjacent normal brain.
- The methylation classifier placed most of these cases in either the desmoplastic infantile ganglioglioma/astrocytoma (DIG/DIA) subgroup or in the infantile hemispheric glioma subgroup.
- For 41 tumors from the intrinsic set in which tissue was available for gene panel and RNA sequencing, 25 tumors had fusions involving either ALK (n = 10), NTRK1 (n = 2), NTRK2 (n = 2), NTRK3 (n = 8), ROS1 (n = 2), or MET (n = 1). BRAF variants (n = 3) were observed in cases that were high scoring by methylation array for the DIG/DIA or DIG/DIA-like subgroups.
- For patients in the intrinsic set, the 5-year survival rate was higher for patients whose tumors had gene fusions when compared with patients whose tumors lacked fusions (approximately 80% vs. 60%, respectively). However, both of these groups of patients had much higher survival rates than other children with high-grade gliomas.
Secondary high-grade glioma
Childhood secondary high-grade glioma (high-grade glioma that is preceded by a low-grade glioma) is uncommon (2.9% in a study of 886 patients). No pediatric low-grade gliomas with the BRAF::KIAA1549 fusion transformed to a high-grade glioma, whereas low-grade gliomas with the BRAF V600E variants were associated with increased risk of transformation. Seven of 18 patients (approximately 40%) with secondary high-grade glioma had BRAF V600E variants, with CDKN2A alterations present in 8 of 14 cases (57%).[24]
Molecular features of glioneuronal and neuronal tumors
Glioneuronal and neuronal tumors are generally low-grade tumors. Select histologies recognized by the 2021 WHO classification include the following:[1]
- Ganglioglioma.
- Desmoplastic infantile ganglioglioma/desmoplastic infantile astrocytoma.
- Dysembryoplastic neuroepithelial tumor.
- Papillary glioneuronal tumor.
- Rosette-forming glioneuronal tumor.
- Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease).
- Gangliocytoma.
- Diffuse leptomeningeal glioneuronal tumor.
- Central neurocytoma.
- Extraventricular neurocytoma.
Ganglioglioma
Ganglioglioma presents during childhood and into adulthood. It most commonly arises in the cerebral cortex and is associated with seizures, but it also presents in other sites, including the spinal cord.[64,65]
The unifying theme for the molecular pathogenesis of ganglioglioma is genomic alterations leading to MAPK pathway activation.[32,66]BRAF alterations are observed in approximately 50% of ganglioglioma cases, with V600E being by far the most common alteration. However, other BRAF variants and gene fusions are also observed. Other less commonly altered genes in ganglioglioma include KRAS, FGFR1, FGFR2, RAF1, NTRK2, and NF1.[32,66]
Desmoplastic infantile astrocytomas (DIA) and desmoplastic infantile gangliogliomas (DIG)
DIA and DIG most often present in the first year of life and show a characteristic imaging appearance in which a contrast-enhancing solid nodule accompanies a large cystic component.[67,68] DIG is more common than DIA,[67] and by methylation array analysis, both diagnoses cluster together.[69] Survival outcome is generally favorable with surgical resection.[67]
The most commonly observed genomic alterations in DIA and DIG are BRAF variants involving V600. Gene fusions involving kinase genes are observed less frequently.
- Among 16 cases confirmed by histology and DNA methylation profiling to be DIA and DIG, BRAF variants were observed in seven cases (43.8%): four BRAF V600E variants and three BRAF V600D variants.[69] One additional case had an EML4::ALK fusion. BRAF variants were present in 4 of 12 DIG cases (25%) (with 3 of 4 altered cases having BRAF V600D) and in 3 of 4 DIA cases (75%) (all 3 altered cases with BRAF V600E).
- One study of seven DIG cases found MAPK pathway alterations in four (57%).[70] Three alterations involved BRAF (V600E, V600D, and one deletion/insertion centered at V600) and one was a TPM3::NTRK1 in-frame fusion. Notably, the variant allele frequency was low (8%–27%), suggesting that DIG is characterized by a prominent nonneoplastic component resulting in low clonal driver variant allele frequencies.
- Another report also described the BRAF V600D variant in a DIG case.[71] As the V600D variant is far less common than V600E in other cancers, its detection in multiple DIG cases suggests an association between the variant and DIG.
Dysembryoplastic neuroepithelial tumor (DNET)
DNET presents in children and adults, with the median age at diagnosis in mid-to-late adolescence. It is characterized histopathologically by the presence of columns of oligodendroglial-like cells and cortical ganglion cells floating in mucin.[72] The temporal lobe is the most common location, and it is associated with drug-refractory epilepsy.[65,73]
FGFR1 alterations have been reported in 60% to 80% of DNETs, and include FGFR1 activating point variants, internal tandem duplication of the kinase domain, and activating gene fusions.[32,74,75]BRAF variants are uncommon in DNET.
Papillary glioneuronal tumor
Papillary glioneuronal tumor is a low-grade biphasic neoplasm with astrocytic and neuronal differentiation that primarily arises in the supratentorial compartment.[35] The median age at presentation is in the early 20s, but it can be observed during childhood through adulthood.
The primary genomic alteration associated with papillary glioneuronal tumor is a gene fusion, SLC44A1::PRKCA, that is associated with the t(9:17)(q31;q24) translocation.[76,77] In one study of 28 cases diagnosed histologically as papillary glioneuronal tumor using methylation arrays, 11 of the cases clustered in a distinctive methylation class, while the remaining cases showed methylation profiles typical for other tumor entities. Molecular analysis of the cases in the distinctive methylation cluster showed that all of them had the SLC44A1::PRKCA gene fusion except for a single case with a NOTCH1::PRKCA gene fusion.[78] This suggests that molecular methods for identifying the presence of a PRKCA fusion are less susceptible to misclassification in diagnosing papillary glioneuronal tumor than are morphology-based methods.
Rosette-forming glioneuronal tumor (RGNT)
RGNT presents in adolescents and adults, with tumors generally located infratentorially, although tumors can arise in mesencephalic or diencephalic regions.[79] The typical histological appearance shows both a glial component and a neurocytic component arranged in rosettes or perivascular pseudorosettes.[35] Outcome for patients with RGNT is generally favorable, consistent with the WHO grade 1 designation.[79]
DNA methylation profiling shows that RGNT has a distinct epigenetic profile that distinguishes it from other low-grade glial/glioneuronal tumor entities.[79] A study of 30 cases of RGNT observed FGFR1 hotspot variants in all analyzed tumors.[79] In addition, PIK3CA activating variants were concurrently observed in 19 of 30 cases (63%). Missense or damaging variants in NF1 were identified in 10 of 30 cases (33%), with 7 tumors having variants in FGFR1, PIK3CA, and NF1. The co-occurrence of variants that activate both the MAPK pathway and the PI3K pathway makes the variant profile of RGNT distinctive among astrocytic and glioneuronal tumors.
Diffuse leptomeningeal glioneuronal tumor (DLGNT)
DLGNT is a rare CNS tumor that has been characterized radiographically by leptomeningeal enhancement on MRI that may involve the posterior fossa, brain stem region, and spinal cord.[80] Intraparenchymal lesions, when present, typically involve the spinal cord.[80] Localized intramedullary glioneuronal tumors without leptomeningeal dissemination and with histomorphological, immunophenotypic, and genomic characteristics similar to DLGNT have been reported.[81]
DLGNT showed a distinctive epigenetic profile on DNA methylation arrays, and unsupervised clustering of array data applied to 30 cases defined two subclasses of DLGNT: methylation class (MC)-1 (n = 17) and MC-2 (n = 13).[80] Of note, many of the array-defined cases had originally been diagnosed as other entities (e.g., primitive neuroectodermal tumors, pilocytic astrocytoma, and anaplastic astrocytoma). Patients with DLGNT-MC-1 were diagnosed at an earlier age than were patients with DLGNT-MC-2 (5 years vs. 14 years, respectively). The 5-year OS rate was higher for patients with DLGNT-MC-1 than for those with DLGNT-MC-2 (100% vs. 43%, respectively). Genomic findings from the 30 cases of methylation array–defined DLGNT are provided below:
- All 30 cases showed loss of chromosome 1p, but only 6 of 17 DLGNT-MC-1 cases showed additional gain of chromosome 1q, compared with all cases of DLGNT-MC-2.[80] A separate report found that chromosome 1q gain was an adverse prognostic factor in patients with DLGNT (including cases with localized disease),[82] which is consistent with the inferior outcome for patients with DLGNT-MC-2.
- Co-deletions of 1p/19q were more frequent in the DLGNT-MC-1 group (7 of 13, 54%) than in the DLGNT-MC-2 group (2 of 13, 15%). In contrast to oligodendroglioma, variants of IDH1 and IDH2 were not identified.[80]
- MAPK pathway activation is common in DLGNT cases.[80] The KIAA1549::BRAF fusion was present in 11 of 15 DLGNT-MC-1 cases (65%) and in 9 of 13 DLGNT-MC-2 cases (69%). Fusions involving NTRK1, NTRK2, or NTRK3 were present in one case each, and another case had a TRIM33::RAF1 fusion.
Extraventricular neurocytoma
Extraventricular neurocytoma is histologically similar to central neurocytoma, consisting of small uniform cells that demonstrate neuronal differentiation. However, extraventricular neurocytoma arises in the brain parenchyma rather than in association with the ventricular system.[35] It presents during childhood through adulthood.
In a study of 40 tumors histologically classified as extraventricular neurocytoma and subjected to methylation array analysis, only 26 formed a separate cluster distinctive from reference tumors of other histologies.[83] Among cases with an extraventricular neurocytoma methylation array classification for which genomic characterization could be performed, 11 of 15 (73%) showed rearrangements affecting members of the FGFR family, with FGFR1::TACC1 being the most common alteration.[83]
Prognosis
Circumscribed astrocytic gliomas, pediatric-type diffuse low-grade gliomas, and glioneuronal/neuronal tumors
These tumors generally carry a relatively favorable prognosis, particularly for well-circumscribed lesions where a radical resection may be possible.[84,85] With the exception of diffuse leptomeningeal glioneuronal tumors, disseminated or multifocal disease is rare.[86]
Unfavorable clinical prognostic features include the following:[87,88,89]
- Young age.
- Inability to obtain a complete resection.
- Diencephalic syndrome.
- Disseminated or multifocal disease. When disseminated or multifocal disease is present, it is associated with a poorer long-term outcome.
On a molecular level, presence of a BRAF V600E variant, especially in conjunction with a CDKN2A or CDKN2B homozygous deletion, has been recognized as a negative prognostic factor, with risk of transformation to a higher-grade tumor. Conversely, the presence of a BRAF::KIAA1549 fusion confers a better clinical outcome in patients with circumscribed astrocytic gliomas.[26][Level of evidence C2]
In children with tumors of the visual pathway, both visual outcomes and clinical assessments are important. Children with isolated optic nerve tumors have a better prognosis than do children with lesions that involve the chiasm or that extend along the optic pathway.[90,91]; [92][Level of evidence C1] Children with NF1 also have a better prognosis, especially when the tumor is found in asymptomatic patients.[93] Better visual acuity at diagnosis, older age at diagnosis, and presence of NF1 are associated with better visual outcomes.[94]
Pediatric-type diffuse high-grade gliomas
These tumors carry a very poor prognosis with currently available therapies.
Patients with diffuse midline glioma, H3 K27-altered have the poorest prognosis, with 3-year survival rates below 5%.[49]
Diffuse brain stem tumors
The following definitions of brain stem tumors are used:
- Brain stem glioma: A general term describing an astrocytoma arising in the brain stem. Such tumors can be circumscribed or diffuse and can occur in any location in the brain stem, including the midbrain, pons, and medulla.
- Diffuse intrinsic pontine glioma (DIPG): A term used to describe an infiltrating astrocytoma (presumed diffuse midline glioma) centered in the pons.
- Diffuse midline glioma, H3 K27-altered: The pathological diagnosis of most tumors that present with imaging features consistent with a DIPG.
The median survival for children with DIPGs is less than 1 year, although about 10% of children will survive longer than 2 years.[95,96] In contrast, patients with focal astrocytomas (e.g., pilocytic astrocytomas) have a markedly improved prognosis, with 5-year OS rates exceeding 90%.[4]
One report from a clinical trial included 42 children and adolescents with newly diagnosed midline thalamic high-grade gliomas. The study found that tumor location, enhancement pattern, diffusion restriction, and variant status did not significantly affect survival.[97] Leptomeningeal metastatic dissemination and lower surgical resection rates were associated with poorer outcomes.
Prognostic factors include the following:
- Histology/grade of the tumor: Astrocytic tumors predominate in the brain stem. WHO grade 1 tumors (e.g., pilocytic astrocytomas and gangliogliomas) have a favorable prognosis and can arise throughout the brain stem, including the tectum of the midbrain, focally within the pons, or at the cervicomedullary junction where they are often exophytic. Low-grade diffuse astrocytomas (WHO grade 2) occurring outside the pons in other brain stem locations tend to be tumors with a more favorable prognosis.[98]
DIPGs are diffuse astrocytomas that, when biopsied at diagnosis, can range from diffuse astrocytomas (WHO grade 2) to glioblastomas (WHO grade 4). At postmortem evaluation, DIPGs are also generally anaplastic astrocytomas (WHO grade 3) or glioblastomas (WHO grade 4) by morphological criteria, although WHO grade 2 regions can also be identified.[52,53,99,100,101]
Approximately 80% of DIPGs, regardless of histological grade, demonstrate a histone H3.3 or H3.1 variant and are now classified by the WHO as diffuse midline gliomas, H3 K27M-altered. All diffuse midline gliomas, H3 K27M-altered, are WHO grade 4, regardless of histological grade, reflecting the poor prognosis of children with this diagnosis.
- Age at diagnosis: Slightly prolonged survival has been found in those either very young (≤3 years) or older (≥10 years) at diagnosis. Approximately 4% of children with DIPGs are diagnosed when younger than 3 years. The prognosis of these children is less dismal than that of older children, with 28% of younger children alive at 2 years compared with 8% of children aged 3 to 10 years at diagnosis and 14% of children older than 10 years at diagnosis. For children aged 10 years and older, long-term survival was associated with older age at presentation and a longer duration of symptoms.[102] The more favorable prognosis for young children may reflect the presence of different biological characteristics in different age groups.[95,103]
- NF1: Children with NF1 and brain stem gliomas may have a better prognosis than other patients who have intrinsic lesions.[104,105]
- Clinical and imaging features present at diagnosis: For children with DIPGs, features associated with surviving less than 2 years include the presence at diagnosis of cranial nerve palsies, ring enhancement, necrosis, and extrapontine extension.[95] The 2-year survival rate is less than 10% for patients with these characteristics.
- Duration of symptoms at diagnosis: Longer duration of symptoms is associated with a more favorable prognosis. The 2-year survival rates range from 7% for patients with duration of symptoms less than 6 months to 29% for patients with duration of symptoms of 24 months or longer.[95]
- Histone variants: Patients with H3.1 K27M variants have a longer median survival (15 months) than do patients with H3.3 K27M variants (10.4 months) or patients without a histone variant (10.5 months).[95]
References:
- Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
- WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
- Kilday JP, Bartels U, Huang A, et al.: Favorable survival and metabolic outcome for children with diencephalic syndrome using a radiation-sparing approach. J Neurooncol 116 (1): 195-204, 2014.
- Klimo P, Pai Panandiker AS, Thompson CJ, et al.: Management and outcome of focal low-grade brainstem tumors in pediatric patients: the St. Jude experience. J Neurosurg Pediatr 11 (3): 274-81, 2013.
- Liu AK, Brandon J, Foreman NK, et al.: Conventional MRI at presentation does not predict clinical response to radiation therapy in children with diffuse pontine glioma. Pediatr Radiol 39 (12): 1317-20, 2009.
- Walker DA, Liu J, Kieran M, et al.: A multi-disciplinary consensus statement concerning surgical approaches to low-grade, high-grade astrocytomas and diffuse intrinsic pontine gliomas in childhood (CPN Paris 2011) using the Delphi method. Neuro Oncol 15 (4): 462-8, 2013.
- Cage TA, Samagh SP, Mueller S, et al.: Feasibility, safety, and indications for surgical biopsy of intrinsic brainstem tumors in children. Childs Nerv Syst 29 (8): 1313-9, 2013.
- Grill J, Puget S, Andreiuolo F, et al.: Critical oncogenic mutations in newly diagnosed pediatric diffuse intrinsic pontine glioma. Pediatr Blood Cancer 58 (4): 489-91, 2012.
- Puget S, Beccaria K, Blauwblomme T, et al.: Biopsy in a series of 130 pediatric diffuse intrinsic Pontine gliomas. Childs Nerv Syst 31 (10): 1773-80, 2015.
- Gupta N, Goumnerova LC, Manley P, et al.: Prospective feasibility and safety assessment of surgical biopsy for patients with newly diagnosed diffuse intrinsic pontine glioma. Neuro Oncol 20 (11): 1547-1555, 2018.
- Pfaff E, El Damaty A, Balasubramanian GP, et al.: Brainstem biopsy in pediatric diffuse intrinsic pontine glioma in the era of precision medicine: the INFORM study experience. Eur J Cancer 114: 27-35, 2019.
- Ryall S, Zapotocky M, Fukuoka K, et al.: Integrated Molecular and Clinical Analysis of 1,000 Pediatric Low-Grade Gliomas. Cancer Cell 37 (4): 569-583.e5, 2020.
- Buczkowicz P, Bartels U, Bouffet E, et al.: Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol 128 (4): 573-81, 2014.
- Holzapfel J, Kandels D, Schmidt R, et al.: Favorable prognosis in pediatric brainstem low-grade glioma: Report from the German SIOP-LGG 2004 cohort. Int J Cancer 146 (12): 3385-3396, 2020.
- Warren KE: Diffuse intrinsic pontine glioma: poised for progress. Front Oncol 2: 205, 2012.
- Packer RJ, Iavarone A, Jones DTW, et al.: Implications of new understandings of gliomas in children and adults with NF1: report of a consensus conference. Neuro Oncol 22 (6): 773-784, 2020.
- D'Angelo F, Ceccarelli M, Tala, et al.: The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat Med 25 (1): 176-187, 2019.
- Franz DN, Agricola K, Mays M, et al.: Everolimus for subependymal giant cell astrocytoma: 5-year final analysis. Ann Neurol 78 (6): 929-38, 2015.
- Mackay A, Burford A, Carvalho D, et al.: Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 32 (4): 520-537.e5, 2017.
- Bouffet E, Larouche V, Campbell BB, et al.: Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol 34 (19): 2206-11, 2016.
- Das A, Tabori U, Sambira Nahum LC, et al.: Efficacy of Nivolumab in Pediatric Cancers with High Mutation Burden and Mismatch Repair Deficiency. Clin Cancer Res 29 (23): 4770-4783, 2023.
- Jones DT, Kocialkowski S, Liu L, et al.: Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68 (21): 8673-7, 2008.
- Hawkins C, Walker E, Mohamed N, et al.: BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res 17 (14): 4790-8, 2011.
- Mistry M, Zhukova N, Merico D, et al.: BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol 33 (9): 1015-22, 2015.
- López GY, Van Ziffle J, Onodera C, et al.: The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol 137 (1): 139-150, 2019.
- Lassaletta A, Zapotocky M, Mistry M, et al.: Therapeutic and Prognostic Implications of BRAF V600E in Pediatric Low-Grade Gliomas. J Clin Oncol 35 (25): 2934-2941, 2017.
- Ho CY, Mobley BC, Gordish-Dressman H, et al.: A clinicopathologic study of diencephalic pediatric low-grade gliomas with BRAF V600 mutation. Acta Neuropathol 130 (4): 575-85, 2015.
- Guerreiro Stucklin AS, Ryall S, Fukuoka K, et al.: Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun 10 (1): 4343, 2019.
- Clarke M, Mackay A, Ismer B, et al.: Infant High-Grade Gliomas Comprise Multiple Subgroups Characterized by Novel Targetable Gene Fusions and Favorable Outcomes. Cancer Discov 10 (7): 942-963, 2020.
- Meredith DM, Cooley LD, Dubuc A, et al.: ROS1 Alterations as a Potential Driver of Gliomas in Infant, Pediatric, and Adult Patients. Mod Pathol 36 (11): 100294, 2023.
- Jones DT, Hutter B, Jäger N, et al.: Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45 (8): 927-32, 2013.
- Qaddoumi I, Orisme W, Wen J, et al.: Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 131 (6): 833-45, 2016.
- Zhang J, Wu G, Miller CP, et al.: Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45 (6): 602-12, 2013.
- Ramkissoon LA, Horowitz PM, Craig JM, et al.: Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci U S A 110 (20): 8188-93, 2013.
- Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.
- Bandopadhayay P, Ramkissoon LA, Jain P, et al.: MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet 48 (3): 273-82, 2016.
- D'Aronco L, Rouleau C, Gayden T, et al.: Brainstem angiocentric gliomas with MYB-QKI rearrangements. Acta Neuropathol 134 (4): 667-669, 2017.
- Chan E, Bollen AW, Sirohi D, et al.: Angiocentric glioma with MYB-QKI fusion located in the brainstem, rather than cerebral cortex. Acta Neuropathol 134 (4): 671-673, 2017.
- Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.
- Lehman NL, Usubalieva A, Lin T, et al.: Genomic analysis demonstrates that histologically-defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol Commun 7 (1): 42, 2019.
- Wood MD, Tihan T, Perry A, et al.: Multimodal molecular analysis of astroblastoma enables reclassification of most cases into more specific molecular entities. Brain Pathol 28 (2): 192-202, 2018.
- Hirose T, Nobusawa S, Sugiyama K, et al.: Astroblastoma: a distinct tumor entity characterized by alterations of the X chromosome and MN1 rearrangement. Brain Pathol 28 (5): 684-694, 2018.
- Lucas CG, Solomon DA, Perry A: A review of recently described genetic alterations in central nervous system tumors. Hum Pathol 96: 56-66, 2020.
- Paugh BS, Qu C, Jones C, et al.: Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol 28 (18): 3061-8, 2010.
- Bax DA, Mackay A, Little SE, et al.: A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin Cancer Res 16 (13): 3368-77, 2010.
- Ward SJ, Karakoula K, Phipps KP, et al.: Cytogenetic analysis of paediatric astrocytoma using comparative genomic hybridisation and fluorescence in-situ hybridisation. J Neurooncol 98 (3): 305-18, 2010.
- Pollack IF, Hamilton RL, Sobol RW, et al.: IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children's Oncology Group. Childs Nerv Syst 27 (1): 87-94, 2011.
- Sturm D, Witt H, Hovestadt V, et al.: Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22 (4): 425-37, 2012.
- Korshunov A, Ryzhova M, Hovestadt V, et al.: Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol 129 (5): 669-78, 2015.
- Castel D, Kergrohen T, Tauziède-Espariat A, et al.: Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol 139 (6): 1109-1113, 2020.
- Rodriguez Gutierrez D, Jones C, Varlet P, et al.: Radiological Evaluation of Newly Diagnosed Non-Brainstem Pediatric High-Grade Glioma in the HERBY Phase II Trial. Clin Cancer Res 26 (8): 1856-1865, 2020.
- Buczkowicz P, Hoeman C, Rakopoulos P, et al.: Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46 (5): 451-6, 2014.
- Taylor KR, Mackay A, Truffaux N, et al.: Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46 (5): 457-61, 2014.
- Auffret L, Ajlil Y, Tauziède-Espariat A, et al.: A new subtype of diffuse midline glioma, H3 K27 and BRAF/FGFR1 co-altered: a clinico-radiological and histomolecular characterisation. Acta Neuropathol 147 (1): 2, 2023.
- Williams EA, Brastianos PK, Wakimoto H, et al.: A comprehensive genomic study of 390 H3F3A-mutant pediatric and adult diffuse high-grade gliomas, CNS WHO grade 4. Acta Neuropathol 146 (3): 515-525, 2023.
- Jain SU, Do TJ, Lund PJ, et al.: PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat Commun 10 (1): 2146, 2019.
- Hübner JM, Müller T, Papageorgiou DN, et al.: EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol 21 (7): 878-889, 2019.
- Chen CCL, Deshmukh S, Jessa S, et al.: Histone H3.3G34-Mutant Interneuron Progenitors Co-opt PDGFRA for Gliomagenesis. Cell 183 (6): 1617-1633.e22, 2020.
- Mackay A, Burford A, Molinari V, et al.: Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell 33 (5): 829-842.e5, 2018.
- Yeo KK, Alexandrescu S, Cotter JA, et al.: Multi-institutional study of the frequency, genomic landscape, and outcome of IDH-mutant glioma in pediatrics. Neuro Oncol 25 (1): 199-210, 2023.
- Suwala AK, Stichel D, Schrimpf D, et al.: Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. Acta Neuropathol 141 (1): 85-100, 2021.
- Reinhardt A, Stichel D, Schrimpf D, et al.: Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol 136 (2): 273-291, 2018.
- Korshunov A, Schrimpf D, Ryzhova M, et al.: H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol 134 (3): 507-516, 2017.
- Becker AJ: Ganglioglioma. In: Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016, pp 138-41.
- Blumcke I, Spreafico R, Haaker G, et al.: Histopathological Findings in Brain Tissue Obtained during Epilepsy Surgery. N Engl J Med 377 (17): 1648-1656, 2017.
- Pekmezci M, Villanueva-Meyer JE, Goode B, et al.: The genetic landscape of ganglioglioma. Acta Neuropathol Commun 6 (1): 47, 2018.
- Bianchi F, Tamburrini G, Massimi L, et al.: Supratentorial tumors typical of the infantile age: desmoplastic infantile ganglioglioma (DIG) and astrocytoma (DIA). A review. Childs Nerv Syst 32 (10): 1833-8, 2016.
- Trehan G, Bruge H, Vinchon M, et al.: MR imaging in the diagnosis of desmoplastic infantile tumor: retrospective study of six cases. AJNR Am J Neuroradiol 25 (6): 1028-33, 2004 Jun-Jul.
- Wang AC, Jones DTW, Abecassis IJ, et al.: Desmoplastic Infantile Ganglioglioma/Astrocytoma (DIG/DIA) Are Distinct Entities with Frequent BRAFV600 Mutations. Mol Cancer Res 16 (10): 1491-1498, 2018.
- Blessing MM, Blackburn PR, Krishnan C, et al.: Desmoplastic Infantile Ganglioglioma: A MAPK Pathway-Driven and Microglia/Macrophage-Rich Neuroepithelial Tumor. J Neuropathol Exp Neurol 78 (11): 1011-1021, 2019.
- Greer A, Foreman NK, Donson A, et al.: Desmoplastic infantile astrocytoma/ganglioglioma with rare BRAF V600D mutation. Pediatr Blood Cancer 64 (6): , 2017.
- Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016.
- Stone TJ, Keeley A, Virasami A, et al.: Comprehensive molecular characterisation of epilepsy-associated glioneuronal tumours. Acta Neuropathol 135 (1): 115-129, 2018.
- Rivera B, Gayden T, Carrot-Zhang J, et al.: Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol 131 (6): 847-63, 2016.
- Matsumura N, Nobusawa S, Ito J, et al.: Multiplex ligation-dependent probe amplification analysis is useful for detecting a copy number gain of the FGFR1 tyrosine kinase domain in dysembryoplastic neuroepithelial tumors. J Neurooncol 143 (1): 27-33, 2019.
- Pages M, Lacroix L, Tauziede-Espariat A, et al.: Papillary glioneuronal tumors: histological and molecular characteristics and diagnostic value of SLC44A1-PRKCA fusion. Acta Neuropathol Commun 3: 85, 2015.
- Bridge JA, Liu XQ, Sumegi J, et al.: Identification of a novel, recurrent SLC44A1-PRKCA fusion in papillary glioneuronal tumor. Brain Pathol 23 (2): 121-8, 2013.
- Hou Y, Pinheiro J, Sahm F, et al.: Papillary glioneuronal tumor (PGNT) exhibits a characteristic methylation profile and fusions involving PRKCA. Acta Neuropathol 137 (5): 837-846, 2019.
- Sievers P, Appay R, Schrimpf D, et al.: Rosette-forming glioneuronal tumors share a distinct DNA methylation profile and mutations in FGFR1, with recurrent co-mutation of PIK3CA and NF1. Acta Neuropathol 138 (3): 497-504, 2019.
- Deng MY, Sill M, Chiang J, et al.: Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol 136 (2): 239-253, 2018.
- Chiang JCH, Harreld JH, Orr BA, et al.: Low-grade spinal glioneuronal tumors with BRAF gene fusion and 1p deletion but without leptomeningeal dissemination. Acta Neuropathol 134 (1): 159-162, 2017.
- Chiang J, Dalton J, Upadhyaya SA, et al.: Chromosome arm 1q gain is an adverse prognostic factor in localized and diffuse leptomeningeal glioneuronal tumors with BRAF gene fusion and 1p deletion. Acta Neuropathol 137 (1): 179-181, 2019.
- Sievers P, Stichel D, Schrimpf D, et al.: FGFR1:TACC1 fusion is a frequent event in molecularly defined extraventricular neurocytoma. Acta Neuropathol 136 (2): 293-302, 2018.
- Wisoff JH, Sanford RA, Heier LA, et al.: Primary neurosurgery for pediatric low-grade gliomas: a prospective multi-institutional study from the Children's Oncology Group. Neurosurgery 68 (6): 1548-54; discussion 1554-5, 2011.
- Bandopadhayay P, Bergthold G, London WB, et al.: Long-term outcome of 4,040 children diagnosed with pediatric low-grade gliomas: an analysis of the Surveillance Epidemiology and End Results (SEER) database. Pediatr Blood Cancer 61 (7): 1173-9, 2014.
- Lu VM, Di L, Gernsback J, et al.: Contemporary outcomes of diffuse leptomeningeal glioneuronal tumor in pediatric patients: A case series and literature review. Clin Neurol Neurosurg 218: 107265, 2022.
- Stokland T, Liu JF, Ironside JW, et al.: A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro Oncol 12 (12): 1257-68, 2010.
- Gnekow AK, Walker DA, Kandels D, et al.: A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer 81: 206-225, 2017.
- Chamdine O, Broniscer A, Wu S, et al.: Metastatic Low-Grade Gliomas in Children: 20 Years' Experience at St. Jude Children's Research Hospital. Pediatr Blood Cancer 63 (1): 62-70, 2016.
- Due-Tønnessen BJ, Helseth E, Scheie D, et al.: Long-term outcome after resection of benign cerebellar astrocytomas in children and young adults (0-19 years): report of 110 consecutive cases. Pediatr Neurosurg 37 (2): 71-80, 2002.
- Massimi L, Tufo T, Di Rocco C: Management of optic-hypothalamic gliomas in children: still a challenging problem. Expert Rev Anticancer Ther 7 (11): 1591-610, 2007.
- Campagna M, Opocher E, Viscardi E, et al.: Optic pathway glioma: long-term visual outcome in children without neurofibromatosis type-1. Pediatr Blood Cancer 55 (6): 1083-8, 2010.
- Hernáiz Driever P, von Hornstein S, Pietsch T, et al.: Natural history and management of low-grade glioma in NF-1 children. J Neurooncol 100 (2): 199-207, 2010.
- Falzon K, Drimtzias E, Picton S, et al.: Visual outcomes after chemotherapy for optic pathway glioma in children with and without neurofibromatosis type 1: results of the International Society of Paediatric Oncology (SIOP) Low-Grade Glioma 2004 trial UK cohort. Br J Ophthalmol 102 (10): 1367-1371, 2018.
- Hoffman LM, Veldhuijzen van Zanten SEM, Colditz N, et al.: Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A Collaborative Report From the International and European Society for Pediatric Oncology DIPG Registries. J Clin Oncol 36 (19): 1963-1972, 2018.
- Cohen KJ, Pollack IF, Zhou T, et al.: Temozolomide in the treatment of high-grade gliomas in children: a report from the Children's Oncology Group. Neuro Oncol 13 (3): 317-23, 2011.
- Rodriguez D, Calmon R, Aliaga ES, et al.: MRI and Molecular Characterization of Pediatric High-Grade Midline Thalamic Gliomas: The HERBY Phase II Trial. Radiology 304 (1): 174-182, 2022.
- McAbee JH, Modica J, Thompson CJ, et al.: Cervicomedullary tumors in children. J Neurosurg Pediatr 16 (4): 357-66, 2015.
- Ballester LY, Wang Z, Shandilya S, et al.: Morphologic characteristics and immunohistochemical profile of diffuse intrinsic pontine gliomas. Am J Surg Pathol 37 (9): 1357-64, 2013.
- Wu G, Diaz AK, Paugh BS, et al.: The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46 (5): 444-50, 2014.
- Hoffman LM, DeWire M, Ryall S, et al.: Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol Commun 4: 1, 2016.
- Erker C, Lane A, Chaney B, et al.: Characteristics of patients ≥10 years of age with diffuse intrinsic pontine glioma: a report from the International DIPG/DMG Registry. Neuro Oncol 24 (1): 141-152, 2022.
- Broniscer A, Laningham FH, Sanders RP, et al.: Young age may predict a better outcome for children with diffuse pontine glioma. Cancer 113 (3): 566-72, 2008.
- Pascual-Castroviejo I, Pascual-Pascual SI, Viaño J, et al.: Posterior fossa tumors in children with neurofibromatosis type 1 (NF1). Childs Nerv Syst 26 (11): 1599-603, 2010.
- Albers AC, Gutmann DH: Gliomas in patients with neurofibromatosis type 1. Expert Rev Neurother 9 (4): 535-9, 2009.