




General Information About Adult Central Nervous System (CNS) Tumors
Incidence and Mortality
Brain tumors account for 85% to 90% of all primary central nervous system (CNS) tumors.[1] Estimated new cases and deaths from brain tumors and other nervous system tumors in the United States in 2024:[2]
- New cases: 25,400.
- Deaths: 18,760.
Data from the Surveillance, Epidemiology, and End Results (SEER) Program database for 2016 to 2020 indicated that the combined incidence of brain and other CNS tumors in the United States was 6.2 per 100,000 people per year, and the mortality rate was 4.4 deaths per 100,000 people per year.[3] Worldwide, approximately 308,102 new cases of brain and other CNS tumors were diagnosed in the year 2020, with an estimated 251,329 deaths.[4]
In general, the incidence of primary CNS tumors is higher in White individuals than in Black individuals, and mortality is higher in men than in women.[3]
Primary brain tumors include the following in decreasing order of frequency:[1]
- Anaplastic astrocytomas and glioblastomas (38% of primary brain tumors).
- Meningiomas and other mesenchymal tumors (27% of primary brain tumors).
- Pituitary tumors.
- Schwannomas.
- CNS lymphomas.
- Oligodendrogliomas.
- Ependymomas.
- Low-grade astrocytomas.
- Medulloblastomas.
Primary spinal tumors include the following in decreasing order of frequency:
- Schwannomas, meningiomas, and ependymomas (79% of primary spinal tumors).
- Sarcomas.
- Astrocytomas.
- Vascular tumors.
- Chordomas.
Primary brain tumors rarely spread to other areas of the body, but they can spread to other parts of the brain and to the spinal axis.
Anatomy
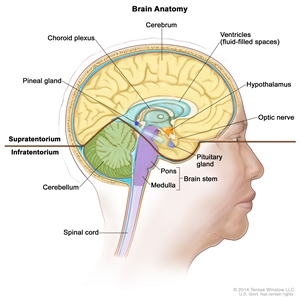
Anatomy of the inside of the brain. The supratentorium contains the cerebrum, ventricles (with cerebrospinal fluid shown in blue), choroid plexus, hypothalamus, pineal gland, pituitary gland, and optic nerve. The infratentorium contains the cerebellum and brain stem.
Risk Factors
Few definitive observations have been made about environmental or occupational causes of primary CNS tumors.[1]
The following potential risk factors have been considered:
- Exposure to vinyl chloride may be a risk factor for glioma.
- Epstein-Barr virus infection has been implicated in the etiology of primary CNS lymphoma.
- Transplant recipients and patients with AIDS have a substantially increased risk of primary CNS lymphoma.[1,5] For more information, see Primary CNS Lymphoma Treatment.
The familial tumor syndromes and related chromosomal abnormalities that are associated with CNS neoplasms include the following:[6,7]
- Neurofibromatosis type 1 (17q11).
- Neurofibromatosis type 2 (22q12).
- von Hippel-Lindau disease (3p25-26).
- Tuberous sclerosis (9q34, 16p13).
- Li-Fraumeni syndrome (17p13).
- Turcot syndrome type 1 (3p21, 7p22).
- Turcot syndrome type 2 (5q21).
- Nevoid basal cell carcinoma syndrome (9q22.3).
Clinical Features
The clinical presentation of various brain tumors is best appreciated by considering the relationship of signs and symptoms to anatomy.[1]
General signs and symptoms include the following:
- Headaches.
- Seizures.
- Visual changes.
- Gastrointestinal symptoms such as loss of appetite, nausea, and vomiting.
- Changes in personality, mood, mental capacity, and concentration.
Seizures are a presenting symptom in approximately 20% of patients with supratentorial brain tumors and may antedate the clinical diagnosis by months to years in patients with slow-growing tumors. Among all patients with brain tumors, 70% with primary parenchymal tumors and 40% with metastatic brain tumors develop seizures at some time during the clinical course.[8]
Diagnostic Evaluation
All brain tumors, whether primary, metastatic, malignant, or benign, must be differentiated from other space-occupying lesions that can have similar clinical presentations, such as abscesses, arteriovenous malformations, and infarctions.[9]
Imaging tests
Contrast-enhanced computed tomography (CT) and magnetic resonance imaging (MRI) have complementary roles in the diagnosis of CNS neoplasms.[1,9,10]
- The speed of CT is desirable for evaluating clinically unstable patients. CT is superior for detecting calcifications, skull lesions, and hyperacute hemorrhages (bleeding less than 24 hours old) and helps direct differential diagnosis and immediate management.
- MRI has superior soft-tissue resolution. MRI can better detect isodense lesions, tumor enhancements, and associated findings such as edema, all phases of hemorrhagic states (except hyperacute), and infarctions. High-quality MRI is the diagnostic study of choice in the evaluation of intramedullary and extramedullary spinal cord lesions.[1]
In posttherapy imaging, single-photon emission computed tomography (SPECT) and positron emission tomography (PET) may be useful in differentiating tumor recurrence from radiation necrosis.[9]
Biopsy
Biopsy confirmation to corroborate the suspected diagnosis of a primary brain tumor is critical, whether before surgery by needle biopsy or at the time of surgical resection. The exception is cases in which the clinical and radiological evidence clearly points to a benign tumor, which could potentially be managed with active surveillance without biopsy or treatment. For other cases, radiological patterns may be misleading, and a definitive biopsy is needed to rule out other causes of space-occupying lesions, such as metastatic cancer or infection.
CT- or MRI-guided stereotactic techniques can be used to place a needle safely and accurately into almost all locations in the brain.
Prognostic Factors
Several genetic alterations have emerged as powerful prognostic factors in diffuse glioma (astrocytoma, oligodendroglioma, mixed glioma, and glioblastoma), and these alterations may guide patient management. Specific alterations include the following:
- DNA methylation of the MGMT gene promoter.
- Mutation of the IDH1 or IDH2 gene.
- Codeletion of chromosomes 1p and 19q.
Other prognostic factors that confer poor prognosis include the following:[11,12]
- Age older than 40 years.
- Progressive disease.
- Tumor size larger than 5 cm.
- Tumor crossing the midline.
- Contrast enhancement on MRI.
- World Health Organization performance status (≥1).
- Neurological symptoms.
- Less than a gross total resection.
In an exploratory analysis of 318 patients with low-grade glioma treated with either radiation therapy alone or temozolomide chemotherapy alone, a combination of these prognostic factors demonstrated the following:[11]
- Longer progression-free survival (PFS) in patients with an IDH mutation without codeletion of 1p/19q when treated with radiation therapy (hazard ratio, 1.86; 95% confidence interval, 1.21–2.87; log-rank, P = .0043).
- No significant treatment-dependent differences in PFS for patients with an IDH mutation with codeletion of 1p/19q and IDH wild-type tumors.
- Patients with wild-type IDH tumors had the worst prognosis independent of treatment type.
- Patients with IDH-mutated tumors with codeletion of 1p/19q had the best prognosis.
- The O6-methylguanine-DNA methyltransferase (MGMT) promoter status in low-grade tumors was methylated in:
- All IDH mutations with codeletion of 1p/19q (45/45).
- Most, but not all (86%, 62/72), of the IDH mutations without codeletion of 1p/19q.
- Fifty-six percent (5/9) of the IDH wild-type cases.
For more information, see the Treatment of Primary Central Nervous System Tumors by Tumor Type section.
References:
- Mehta M, Vogelbaum MA, Chang S, et al.: Neoplasms of the central nervous system. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Lippincott Williams & Wilkins, 2011, pp 1700-49.
- American Cancer Society: Cancer Facts and Figures 2024. American Cancer Society, 2024. Available online. Last accessed June 21, 2024.
- National Cancer Institute: SEER Cancer Stat Facts: Brain and Other Nervous System Cancer. Bethesda, Md: National Cancer Institute. Available online. Last accessed March 5, 2024.
- Sung H, Ferlay J, Siegel RL, et al.: Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 71 (3): 209-249, 2021.
- Schabet M: Epidemiology of primary CNS lymphoma. J Neurooncol 43 (3): 199-201, 1999.
- Behin A, Hoang-Xuan K, Carpentier AF, et al.: Primary brain tumours in adults. Lancet 361 (9354): 323-31, 2003.
- Kleihues P, Cavenee WK, eds.: Pathology and Genetics of Tumours of the Nervous System. International Agency for Research on Cancer, 2000.
- Cloughesy T, Selch MT, Liau L: Brain. In: Haskell CM: Cancer Treatment. 5th ed. WB Saunders Co, 2001, pp 1106-42.
- Hutter A, Schwetye KE, Bierhals AJ, et al.: Brain neoplasms: epidemiology, diagnosis, and prospects for cost-effective imaging. Neuroimaging Clin N Am 13 (2): 237-50, x-xi, 2003.
- Ricci PE: Imaging of adult brain tumors. Neuroimaging Clin N Am 9 (4): 651-69, 1999.
- Baumert BG, Hegi ME, van den Bent MJ, et al.: Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 17 (11): 1521-1532, 2016.
- Reijneveld JC, Taphoorn MJ, Coens C, et al.: Health-related quality of life in patients with high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 17 (11): 1533-1542, 2016.