




When a genetic explanation for a neurological disorder is imprecise or there is a suspected genetic cause, neurologists can now call on the Neurogenetics Program recently launched by the Department of Neurology at NewYork-Presbyterian/
Matthew B. Harms, MD, is a clinician-scientist in neurophysiology and neuromuscular medicine and Associate Professor of Neurology at Columbia. A member of the Eleanor and Lou Gehrig ALS Center in the Department of Neurology at NewYork-Presbyterian/
As a pediatric and neonatal neurologist and genetic epileptologist Tristan T. Sands, MD, PhD, faces similar situations in his clinical practice and research endeavors, which focus on seizures, epilepsies, and developmental and epileptic encephalopathies resulting from genetic causes. “Over the last 20 years, research has demonstrated that there are many epilepsies. Knowing the genetic underpinnings of someone’s epilepsy means, for the first time, that we are really able to understand the nature of their condition,” says Dr. Sands, an Assistant Professor of Neurology at Columbia. “Epilepsy can be caused by a number of different factors, but it has two peaks of onset. One in childhood, one later on in life. We believe that there is pronounced genetic influence especially in epilepsy that first presents during childhood.”
“The obvious place to start when thinking about genetics are diseases that present in young patients,” says Dr. Harms. “There is a much higher likelihood that a genetic disorder that manifests before puberty will have a genetic etiology. It would then seem to follow that there may be older individuals with milder forms of that genetic disorder whose symptoms didn’t present until adulthood. Lo and behold, when you sequence those same genes in adults, you find that is indeed the case. At Columbia, we have forged closer relationships between pediatric neurology and adult neurology because so many of these disorders span the age spectrum and we now know they are linked, in some cases, by a genetic causation.”
“As Tristan notes about epilepsy, the same type of genetic mutations that appear in infants with severe seizures have been found in adults with relatively mild forms of epilepsy,” adds Dr. Harms. “That’s also the case for the inherited neuromuscular diseases such as neuropathy, myopathy, spinal muscular atrophy, and any number of diseases that present in childhood in which milder forms of the disease caused by the same genes appear in young adults in their 20s and 30s, and in some cases, up into middle and late ages. So we think of neurogenetics as a field that transverses generations. Many of our clinicians have overlapping practices. Tristan is a pediatric neurologist who sees adults with epilepsy. And while I'm an adult neurologist, I also see patients in the pediatric neuromuscular clinic.”
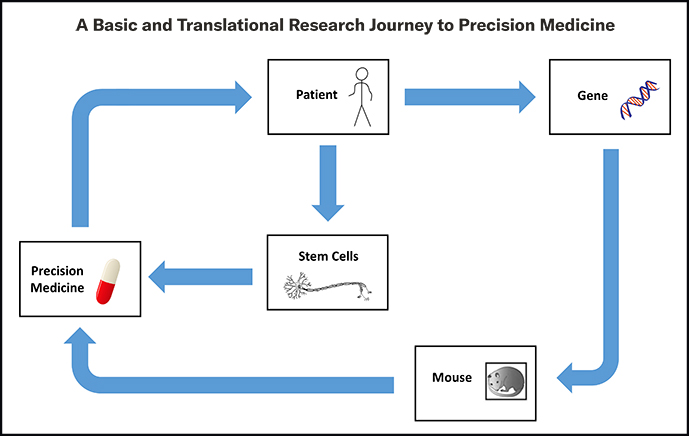
Neurogenetics Program: Filling the Gap When the Genetic Link is Elusive
Dr. Harms, Dr. Sands, and their colleagues in the adult neurology programs at NewYork-Presbyterian/
“The Neurogenetics Program at Columbia leverages the unrivaled genetics expertise and latest sequencing technologies to be able to delve into cases in which a clear diagnosis or genetic association has not been identified, at times using technologies that are not readily available elsewhere for this type of investigation,” says Dr. Harms. “This sets us apart from other genetic programs and could be of interest to neurologists in the community who do not have the resources of an academic medical center when encountering a patient who may have a genetic mutation. We are anticipating that doing this type of research today will lead to the discovery of new genes and hopefully molecular therapy in five years’ time.”
For physicians and patients, the novel neurogenetics program offers a number of benefits. For physicians, the program has the expertise, the tools, and the latest generation sequencing options to evaluate and comprehensively test patients who remain in limbo without a diagnosis and also serves as a source of information for future research endeavors. For patients, identifying a genetic etiology for their neurological disease that conventional genetic technologies did not reveal can help end their “diagnostic odyssey” that is often faced by those with undiagnosed conditions. The program also provides opportunities for targeted treatment and/or improved medical management and offers the potential for enrollment into gene-specific clinical and research trials.
Dr. Harms and Dr. Sands are also on the faculty of Columbia’s prestigious Institute for Genomic Medicine (IGM), which collaborates with clinical departments to provide a comprehensive interpretation of patient genomes and is a resource for NewYork-Presbyterian clinicians to make effective use of genomic data to improve patient care. The IGM specifically targets therapeutic areas, including ALS and epilepsy, in which genomic data is expected to provide an important clinical impact. The Institute’s advanced precision medicine approaches incorporate studies of molecular and small molecule therapies directed against the mutations identified in patients.
In collaboration with the Institute for Genetic Medicine, Dr. Harms' laboratory is using whole genome and transcriptome sequencing to bring precision medicine to amyotrophic lateral sclerosis, while Dr. Sands is evaluating patients with presumed genetic etiologies and conducting research on precision medicine for the genetic epilepsies.
Our Neurogenetics Program sets us apart from other genetic programs and could be of interest to neurologists in the community who do not have the resources of an academic medical center when encountering a patient who may have a genetic mutation.
For physicians and patients, the novel neurogenetics program offers a number of benefits. For physicians, the program has the expertise, the tools, and the latest generation sequencing options to evaluate and comprehensively test patients who remain in limbo without a diagnosis and also serves as a source of information for future research endeavors. For patients, identifying a genetic etiology for their neurological disease that conventional genetic technologies did not reveal can help end their “diagnostic odyssey” that is often faced by those with undiagnosed conditions. The program also provides opportunities for targeted treatment and/or improved medical management and offers the potential for enrollment into gene-specific clinical and research trials.
Dr. Harms and Dr. Sands are also on the faculty of Columbia’s prestigious Institute for Genomic Medicine (IGM), which collaborates with clinical departments to provide a comprehensive interpretation of patient genomes and is a resource for NewYork-Presbyterian clinicians to make effective use of genomic data to improve patient care. The IGM specifically targets therapeutic areas, including ALS and epilepsy, in which genomic data is expected to provide an important clinical impact. The Institute’s advanced precision medicine approaches incorporate studies of molecular and small molecule therapies directed against the mutations identified in patients.
In collaboration with the Institute for Genetic Medicine, Dr. Harms' laboratory is using whole genome and transcriptome sequencing to bring precision medicine to amyotrophic lateral sclerosis, while Dr. Sands is evaluating patients with presumed genetic etiologies and conducting research on precision medicine for the genetic epilepsies.
Amyotrophic Lateral Sclerosis: Genetic Understanding Coming Into its Own
Of the estimated 15,000 individuals with amyotrophic lateral sclerosis (ALS) in the United States about 10 to 15 percent have a mutation in one of more than 30 genes associated with the disease that cause the production of proteins toxic to motor neurons.

Dr. Matthew Harms
In 2011, Dr. Harms and his colleagues were the first to use next generation sequencing technologies to comprehensively investigate the genetic landscape of ALS. They also began exploring genetic influences on C9ORF72 ALS disease phenotypes. Dr. Harms directed the clinical phenotyping effort of a large multicenter collaboration that in 2015 identified TBK1 as a novel ALS gene and highlighted several candidate genes for further studies.
Most recently, Dr. Harms, Neil Shneider, MD, PhD, Director of the Eleanor and Lou Gehrig ALS Center at Columbia, and their colleagues in the ALS Center launched Silence ALS, a new initiative to develop experimental personalized therapies that focus on patients with the rarest of ALS gene mutations. Each mutation is thought to affect between 1 and 30 people worldwide.
Individuals will be identified through Columbia’s ALS Families Project, a study established in 2018 and directed by Dr. Shneider and Dr. Harms that follows pre-symptomatic carriers of ALS-associated gene mutations with the goal of developing markers of ALS that can guide clinical efforts to intervene early in the course of the disease.
Silence ALS aims to treat patients early in the course of their disease, ideally before onset of symptoms, with antisense oligonucleotides (ASOs) – short interfering pieces of DNA or RNA – that bind to RNA and prevent it from making the toxic proteins. Some ASOs are currently undergoing testing in people with more common genetic forms of ALS, but these therapies are not available for people with unique or extremely rare mutations.
“With the goal of developing ASOs that are specific to each patient and beginning treatment before symptoms appear, Silence ALS will provide the infrastructure to rapidly identify patients with very rare ALS mutations and design therapies that may delay or prevent disease onset or slow its progression once it has begun,” says Dr. Shneider, the project’s principal investigator.
“Silence is a reference to using genetic therapies to turn off genes that we think are driving the disease,” explains Dr. Harms. “This is very much a precision medicine initiative. We perform ALS gene sequencing in people who are diagnosed with the disease. When we find a causative mutation in one of the ALS genes, we assess whether the mutated allele is amenable to silencing using any one of the molecular tools that we have available. These would include custom-designed RNA-based therapies with ASOs to try to shut off the bad copy of the gene. It’s very exciting, and we are leaders in the world in pioneering these types of approaches. Importantly, our ability to identify these mutations and to develop next generation molecular therapies hinge on the robust clinical infrastructure for genetic sequencing and discovery that we have here.”
Silence ALS is a direct outgrowth of the work by the Columbia team to design a similar molecule for the FUS gene, which is now in clinical trials. That experimental drug – jacifusen – was shown to lower levels of FUS, a toxic protein, in an animal model and the hope is that it may alter the progression of ALS. “With jacifusen, we have already implemented the precision medicine promise,” says Dr. Harms. “We are now in the process of scaling up our efforts from the more common gene to more specific gene mutations that affect a small subset of patients with ALS or other neuromuscular diseases.”
In parallel with Silence ALS, the ALS Center established PREVENT ALS, a network to unify programs that offer genetic counseling and decision-making strategies for at risk relatives who may want to know whether they carry an ALS gene mutation. “The ultimate goal of PREVENT ALS, and the programs it brings together, is to identify people who are at risk for developing ALS because they carry gene mutations that cause it, but don't yet have symptoms,” says Dr. Harms. “Through this program, in addition to identifying potential candidates for therapy trials down the road, we are also doing biomarker discovery. We still need a reliable marker of symptom conversion in the presymptomatic population. At this point, we can only offer retrospective analysis using standard clinical testing while waiting to see if the person develops symptoms. However, the drug developed for the individual who has the disease will be the same drug used for their relatives who carry the gene mutation but are currently presymptomatic or have mild symptoms. Once we establish the safety of an antisense oligonucleotide, for example, we would definitely consider using it in presymptomatic individuals as a preventative measure.”
Dr. Harms is also wrapping up a study called Genomic Translation for ALS Care (GTAC), in which Columbia researchers sequenced the genomes of 1,400 patients with ALS and followed them for up to three years to ascertain how quickly their disease progressed. The study also looked at potential environmental and lifestyle factors that could trigger the onset of symptoms in individuals with the gene mutation. “Because ALS is very clinically heterogeneous as are the lifestyle and environmental backgrounds of our patients, unless there is a very strong environmental influence that trumps all others, the likelihood of detecting an associated external factor in a cohort of this size is relatively small. But it’s a start,” says Dr. Harms.
Epilepsy: Unveiling the Influence of Genetics on Etiology
As with Dr. Harms’ genetics research in neuromuscular disease, Dr. Sands is pursuing research involving gene sequencing related to epilepsy in both children and adults to identify those patients who have a genetic diagnosis. In a genetic screening for epilepsy conducted by Columbia’s Institute for Genomic Medicine, the yield of exome across a broad unselected pediatric and adult cohort was approximately 10 percent. Two consistent patterns were seen: Comorbidities reflecting other forms of cerebral dysfunction, i.e., encephalopathy, and earlier age of onset, especially onset in the first years of life, were both associated with a higher yield. Only 10 genes were found in more than one individual and 48 different genes were found overall.

Dr. Tristan Sands
“Epilepsy is a predisposition to unprovoked seizures, but there is a vast spectrum of what this means,” says Dr. Sands. “Historically, it was generally accepted in the field that there was some genetic influence over epilepsy. For instance, higher rates of epilepsy were observed in identical twins compared to fraternal twins. There were higher rates of epilepsy in first-degree relatives than in the general population. However, it wasn’t until the late 1990s that a number of families with rare, dominantly inherited epilepsy syndromes were mapped, leading to the initial discovery of a gene, that when changed, resulted in epilepsy. In these cases, the gene was being passed down in an autosomal dominant fashion through families.”
With the advent of next generation sequencing, a second, much larger wave of epilepsy gene discovery came to fruition in the 2000s and continues to this day. “The high throughput gene sequencing approach allowed for exploration of the genetics of sporadic epilepsy,” notes Dr. Sands. “Since this type of epilepsy was clearly not inherited, research focused on uncovering a new genetic change to account for the epilepsy. That resulted in the identification of more than 80 genes strongly associated with epilepsy and hundreds more that cause epilepsy as a variable aspect of a more complex phenotype.” Columbia’s Institute for Genomic Medicine has been instrumental in characterizing a number of these genes, including CSNK2B, KCNQ3, GNB1, and NBEA, for which Dr. Sands is a key collaborator. The IGM’s basic and translational research continues to uncover mechanisms within these and other genes toward the ultimate goal of developing new treatments.
The complexity of extricating the genetic contributions to the etiology of epilepsy is staggering in light of its genotypic and phenotypic heterogeneity. Similar epilepsy can arise from variants in more than one gene, and variants in the same gene can give rise to more than one epilepsy.
“While epilepsy often starts in childhood, we found that those same genetic-based epilepsies can present in adulthood,” says Dr. Sands. “I think the significance of this is underappreciated. Genetic associations with epilepsy in the adult population are not as front and center as they are in the pediatric epilepsy world and therefore may go under-recognized by providers caring for adult patients with epilepsy. As more information becomes available about the different genes associated with epilepsy, efforts are also underway to identify treatments that target the underlying etiology – essentially precision medicine for single gene causes of epilepsy. But you have to have the genetic diagnosis to be considered for such treatment.”
The challenge that remains, notes Dr. Sands, is who do you test since most of the epilepsies are not inherited or clustered with any particular ancestral background, race or ethnicity, but rather they occur spontaneously. What features of epilepsy are associated with a genetic diagnosis? Currently, Dr. Sands does not recommend testing for patients with idiopathic epilepsy syndromes unless a comorbid neurodevelopmental disability is present or onset is prior to three years of age and generally, with some caveats, for individuals with epilepsy that is apparently acquired.
“Some of the variants that we’re talking about are not large-scale chromosomal changes,” says Dr. Sands. “Sometimes, just changing one amino acid in one important protein is sufficient to trigger the epilepsy. Ultimately, our focus is understanding the genetic landscape of epilepsy in order to improve patient care. The most exciting way that we foresee this happening is through precision medicine with disease modifying treatments such as antisense oligonucleotides and small molecules – several which are now in development – that target the particular genetic cause of epilepsy.”

