




Scientists from NewYork-Presbyterian have unraveled a 40-year mystery about the mechanism through which the heart contracts in response to danger. After two decades of investigation, the researchers deciphered the fundamental molecular drivers of cardiac contractility during the fight-or-flight response, and identified a novel therapeutic target that could lead to more effective treatments for people with heart failure. The findings were published as a cover story in the November 2022 issue of Nature Cardiovascular Research.

Dr. Steven Marx
When a person senses danger, the surge in adrenaline stimulates beta-adrenergic receptors—proteins on the surface of heart cells called cardiomyocytes—which modulate the influx of calcium ions into the heart via calcium channels to initiate cardiac contractility. Just how this happens, however, has been a topic of debate since the 1970s. "Many scientists have proposed various theories to explain the mechanism behind this reaction, all of which were proven in later studies not to be correct," said the study's senior author, Steven O. Marx, MD, a cardiologist at NewYork-Presbyterian/
Utilizing a genetically modified mouse model they created, Dr. Marx and his colleagues identified the protein Rad as the major regulator of cardiac contractility in response to adrenaline. Rad acts as a brake on the activity of calcium channels, but adrenaline releases this brake. During the fight-or-flight response, Rad releases from the channel, allowing for an increase in cardiac contraction and giving us the boost of heart contraction we need to flee a predator, for example. "This process has evolved over millions and millions of years to escape danger," Dr. Marx explained. "We found that when the sympathetic nervous system is activated and Rad releases from the calcium channels, additional calcium channels are recruited that would have normally been silent."
We discovered a new mechanism behind regulation of the calcium channel, the fundamental process that causes the heart to contract during fight or flight.
— Dr. Steven O. Marx
At the heart of this process is a protein called protein kinase A (PKA), which activates the calcium channel in the presence of adrenaline. For years, it was thought that PKA directly alters specific regions within the channel called PKA phosphorylation sites, but this theory subsequently proved incorrect. In a seminal paper published in Nature in January 2020, Dr. Marx and his team reported their findings using mice genetically engineered to have cardiomyocytes lacking PKA phosphorylation sites within the calcium channels. When the cardiomyocytes were stimulated with an adrenaline-like drug, the calcium channels continued to respond, suggesting an unknown factor involved in the process.

An illustration of the cardiac fight-or-flight response
(Credit: Brian Soda/
Collaborating with scientists from Harvard Medical School, the researchers used proximity proteomics: a technique in which a protein of interest is fused to an enzyme that tags nearby proteins, which can then be identified by mass spectrometry. They identified virtually every protein near calcium channels in both mouse cardiomyocytes and intact mouse hearts before and after exposure to the adrenaline-like drug. They found that Rad was the only protein that consistently demonstrated a significant change in levels after exposure to adrenaline. It was enriched under resting conditions but depleted when beta-adrenergic receptors were stimulated. PKA phosphorylates Rad, inducing it to leave the area around a calcium channel and preventing it from inhibiting the calcium channels.
Dr. Marx and his fellow investigators—including Geoffrey S. Pitt, MD, PhD, Ida and Theo Rossi Distinguished Professor of Medicine and Director of the Cardiovascular Research Institute at NewYork-Presbyterian/
They confirmed that phosphorylation of Rad by PKA was indeed essential for the sympathetic nervous system stimulation of calcium channels and for the heart to increase contractility when stimulated by adrenaline. On the other hand, preventing Rad from binding to calcium channels increased contractility. "The finding that this process depends almost exclusively on the phosphorylation of Rad by PKA was unexpected," noted Dr. Marx.
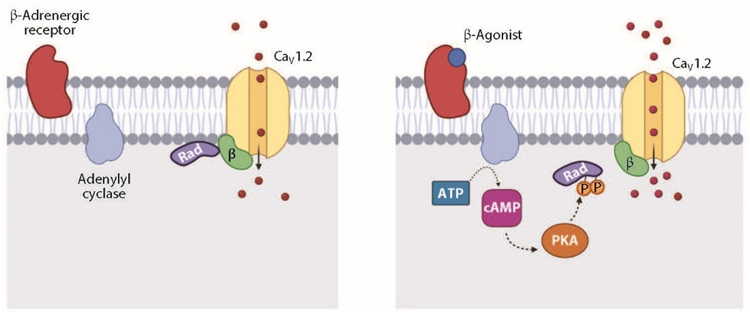
Phosphorylation of Rad is required for cAMP-PKA activation of calcium channels. PKA phosphorylates several residues on Rad, causing dissociation of Rad from the calcium channels and increased calcium ion influx.
What does this mean for patients with heart failure, who experience reduced cardiac contractility? Currently, medications used in the critical care setting to boost the heart's pumping ability—such as the beta-adrenergic agonist dobutamine and the phosphodiesterase inhibitor milrinone—can cause arrhythmias, hypotension, and tachycardia because they target the entire adrenergic system. By identifying Rad as a key player in heart contractility, scientists have an opportunity to develop novel therapeutics that take aim at this component of the signaling cascade, which may lead to more potent and effective therapies for the failing heart, with fewer side effects. Dr. Marx and his team have received a Columbia Translational Therapeutics Accelerator pilot award from the Irving Institute for Clinical and Translational Research and Columbia Technology Ventures to carry out the initial phases of the drug development program.
By identifying Rad as a key player in heart contractility, scientists have an opportunity to develop novel therapeutics for people with heart failure.
The study has been praised by others in the field. "The work presented here represents a major advance in our mechanistic understanding of how the heart functions. Overall, the authors have clarified more precisely the mechanism underlying the inotropic reserve of the heart. A therapeutic regimen based on this work would represent a novel treatment for heart failure," wrote Walter Koch, PhD, Chair of Cardiovascular Sciences at Temple University in an accompanying expert opinion.
"The roots of this process have been a mystery, and we like to solve a mystery. First people said it was solved, and then they said it was unsolvable," said Dr. Marx. "The next step is to develop a new class of much-needed molecules or gene therapy to enhance cardiac contractility in people with heart failure. This intervention has great therapeutic potential because it bypasses the beta-adrenergic signaling system entirely and only targets Rad, the key downstream regulator of cardiac contractility."
